02 Apr 2024
Ph.D. Dissertation Defense by Zhe Wang,
- Tues., 04/02/24, at 12:00PM (ET)
Committee:
Matthew Dwyer, Committee Chair (CS/SEAS/UVA)
Yanjun Qi, Advisor (CS/SEAS, SDS, SM/UVA)
Miaomiao Zhang (ECE/CS/SEAS/UVA)
Jianhui Zhou (Statistics/College/UVA)
Vicente Ordonez (CS/Rice University)
Title: Toward Out-Of-Distribution Generalization Of Deep Learning Models
- Abstract : Deep learning models, especially deep neural networks (DNNs), perform extremely well when the testing and training distributions align. However, real-world scenarios often witness shifts in data distribution across domains, and tasks, over time, and are influenced by adversarial attacks. Such shifts from the training to testing distribution present challenges, resulting in performance degradation of DNNs. The varied testing distributions from diverse users underscore the urgent necessity to understand OOD problems and design methods to mitigate OOD generalization challenges. Therefore, this dissertation develops methods and strategies to enhance DNNs’ ability to generalize to unseen distributions. First, we focus on generalizing DNNs to unknown domains, in which no prior information about testing domains is available during training. We propose a novel optimization approach that learns principal gradients from eigenvectors of training optimization trajectories. This robust gradient design forces the training to ignore domain-dependent noise signals and updates all training domains with a robust direction covering the main components of parameter dynamics. Second, we focus on designing strategies to generalize DNNs to unseen tasks (i.e. meta-learning), for instance, a new unknown RL task with few demonstration trajectories. The main challenge is to infer the potential identity of a new task from a limited number of annotated samples. We propose modeling a new task’s identity as a stochastic variable and encoding it with a stochastic neural network. This task identity design helps meta-learning to adapt shared training knowledge to a new current task. When solving similar task generalization issues in offline RL, we further propose learning from the RL transition dynamic and reward function to capture a task’s identity. Third, deep learning models should not only perform well on clean, legitimate data distribution but also on data that has been subjected to adversarial attacks. Entering the era of large foundation models, we focus on techniques to craft adversarial attackers for jailbreaking pretrained large language models (LLMs) due to their prevalent recent adoptions. We design a new objective, which learns adversarial suffixes with much cheaper queries and higher attack success rate. The learned suffixes also demonstrate higher transferability across LLMs. In the thesis, we validate the effectiveness of our methods across image classification and completion, wealth index regression from satellite images, robotic control, real-world temperature forecasting, and natural language generation.
29 Jun 2022
Arshdeep Sekhon’s PhD Defense
Committee:
Yanjun Qi (Advisor)(CS/SEAS/UVA)
Matthew Dwyer (CS/SEAS/UVA)
Yangfeng Ji (CS/SEAS/UVA)
Vicente Ordonez (CS/SEAS/UVA)
Jianhui Zhou (Department of Statistics/UVA)
Title: Relational Structure Discovery for Deep Learning
- Abstract:
Graph structure is ubiquitous: from physical relationships to biological interactions to social networks, and many more spread across the universe. Not only is the world around us rich in relational structure, but our mental model of the world is also structured: we think, reason, and communicate in terms of entities and their relations. Such a graph-structured real world calls for artificial intelligence methods that think like humans and hence employ this structure for decision making. Realizing such a framework requires known structure/graph and models that can ingest these non-linear graphical inputs. In cases of a latent unknown graph structure, state-of-the-art deep learning models either focus on task-agnostic statistical dependency learning or diverge from explicit feature dependencies during prediction. We bridge this gap and introduce methods for jointly learning and incorporating graph-based relational knowledge into state-of-the-art deep learning models to help improve (1) predictions, (2) interpretability, (3) post-hoc interpretations, and (4) test datasets selection. Specifically, we contribute methods that enable learning graphical relationships from data without such a ground truth graph. Furthermore, we introduce plug-and-play methods that bias deep learning models to include the learned graph explicitly for improving the aforementioned downstream tasks. We demonstrate our methods’ capabilities on simulated, tabular, NLP, and vision tasks.
20 Jul 2021
Ph.D. Dissertation Defense by Jack Lanchantin
- Tuesday, July 20th, 2021 at 2:00 PM (ET), via Zoom.
Committee:
- Vicente Ordóñez Román, Committee Chair, (CS/SEAS/UVA)
- Yanjun Qi, advisor, (CS/SEAS/UVA)
- Yangfeng Ji (CS/SEAS/UVA)
- Clint Miller (Public Health Sciences/SOM/UVA)
- Casey Greene (Biochemistry & Molecular Genetics/SOM/University of Colorado)
Title: Modeling interactions with Deep Learning
- Abstract: Interacting systems are highly prevalent in many real-world settings, including genomics, proteomics, and images. The dynamics of complex systems are often explained as a composition of entities and their interaction graphs. In this dissertation, we design state-of-the-art deep neural networks for interaction-oriented representation learning. Learning such structure representations from data can provide semantic clarity, ease of reasoning for generating new knowledge, and potentially causal interpretation. We consider three different types of interactions: 1) interactions within a particular input sample, 2) interactions between multiple input samples, and 3) interactions between output labels. For each type of interaction, we design novel models to tackle a real-world problem and validate our results both quantitatively and visually.
01 Jul 2021
I gave a tutorial talk at UVA-VADC Seminar Series 2021 and at monthly NIH Data Science Showcase seminar.
Title: Make Deep Learning Interpretable for Sequential Data Analysis in Biomedicine
This tutorial includes four of our recent papers:
Thanks for reading!
11 Jun 2021
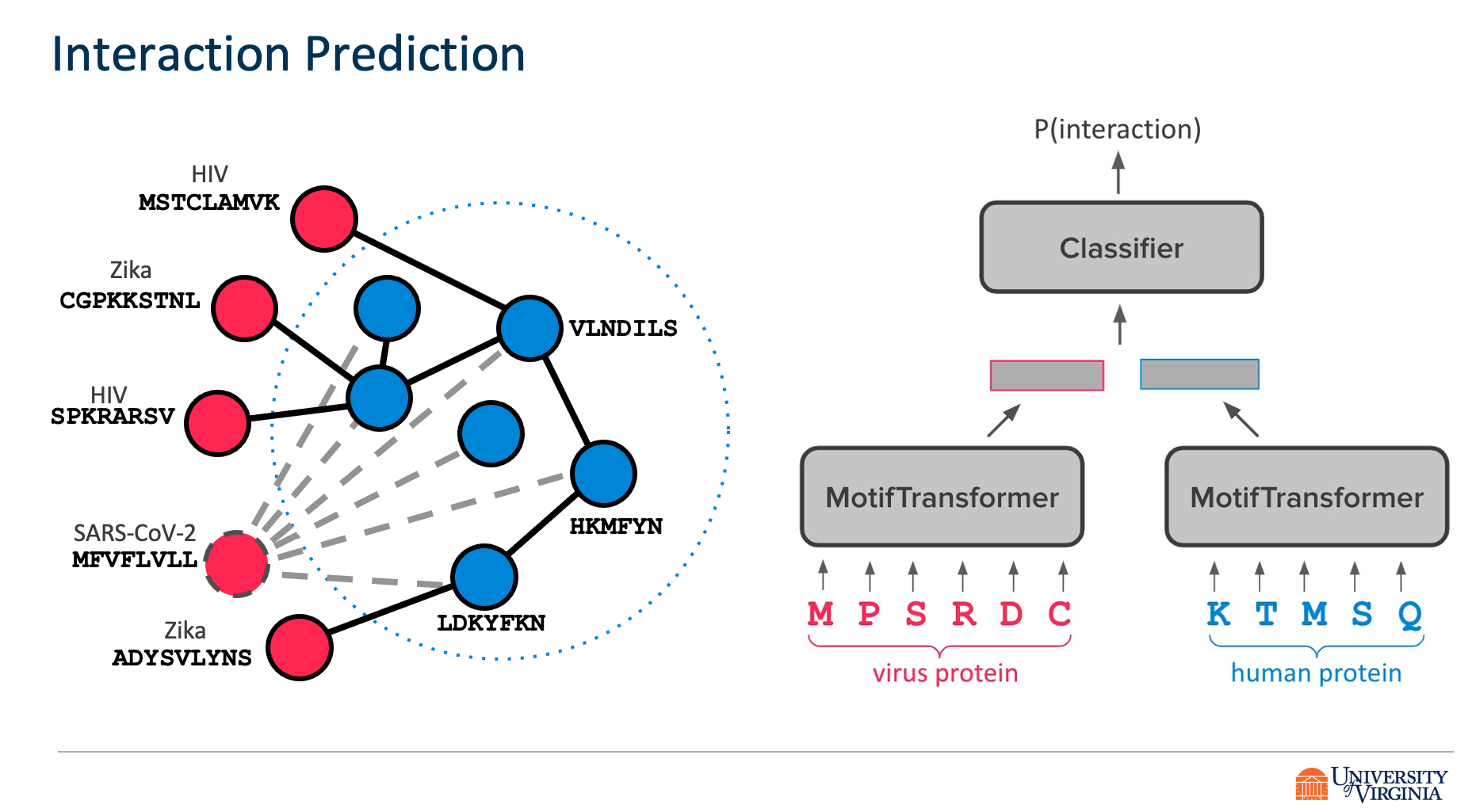
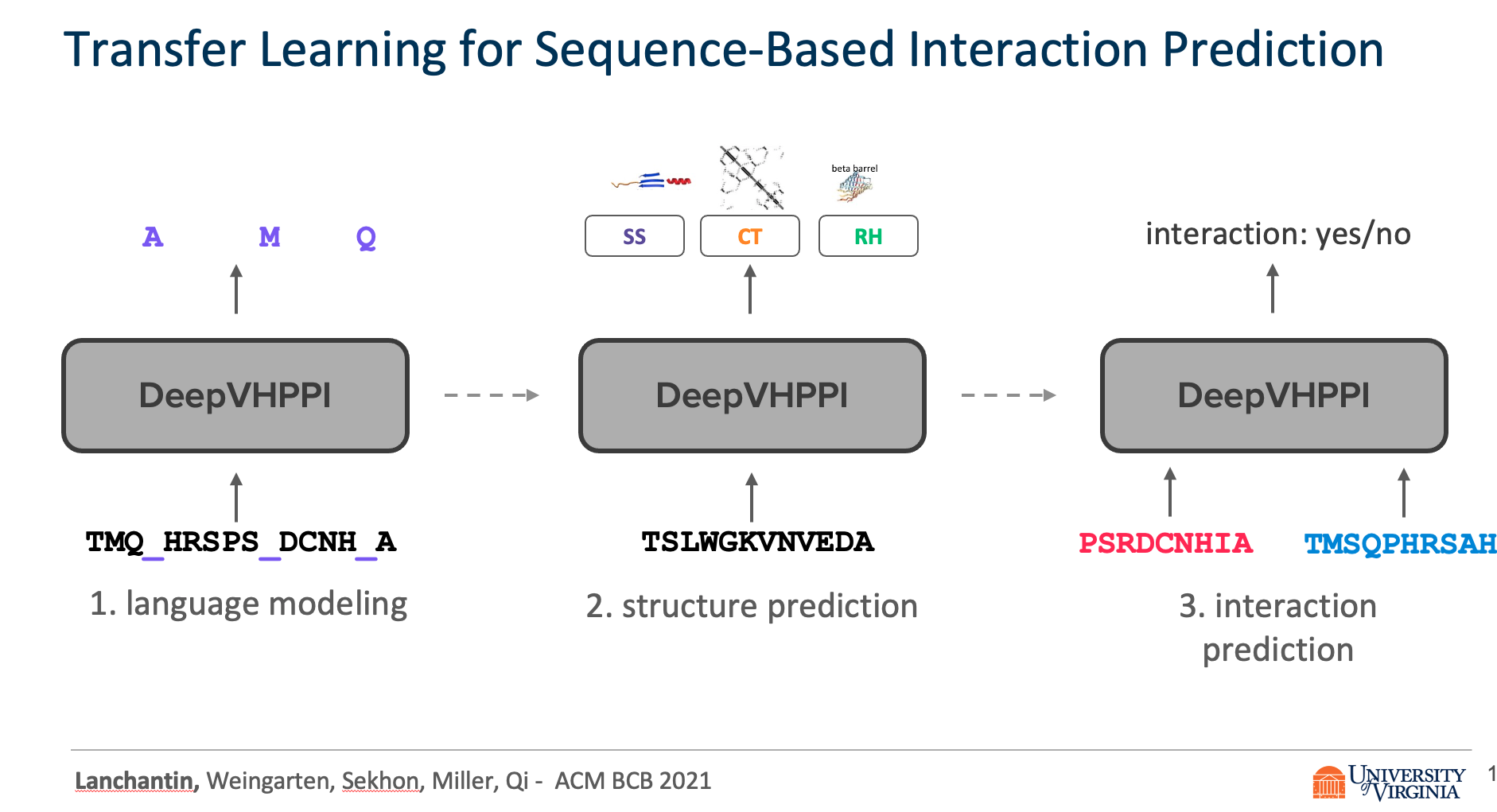
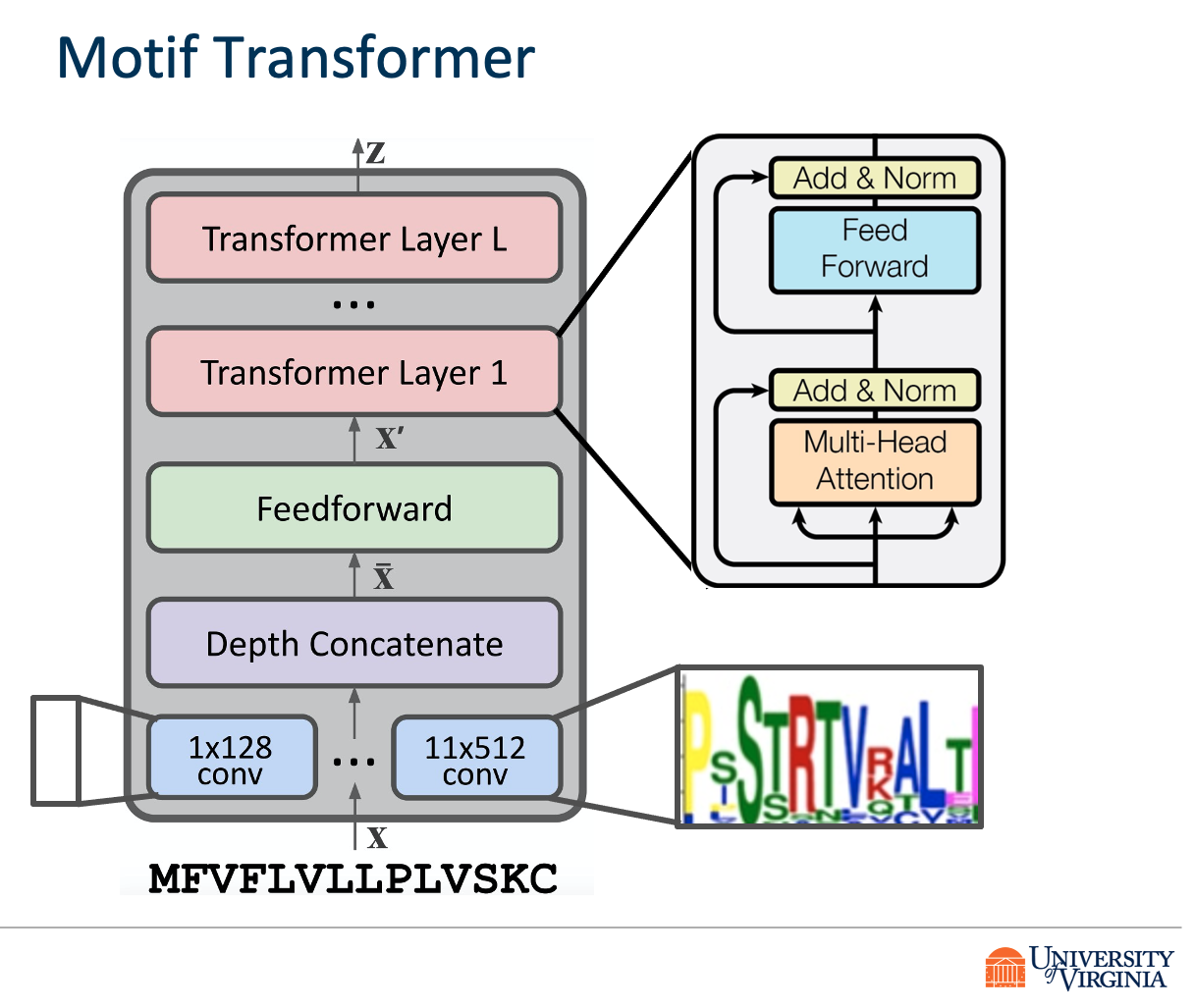
Title: Transfer Learning for Predicting Virus-Host Protein Interactions for Novel Virus Sequences
- authors: Jack Lanchantin, Tom Weingarten, Arshdeep Sekhon, Clint Miller, Yanjun Qi
- 2021 ACM Conference on Bioinformatics, Computational Biology, and Health Informatics (ACM BCB)
Abstract
Viruses such as SARS-CoV-2 infect the human body by forming interactions between virus proteins and human proteins. However, experimental methods to find protein interactions are inadequate: large scale experiments are noisy, and small scale experiments are slow and expensive. Inspired by the recent successes of deep neural networks, we hypothesize that deep learning methods are well-positioned to aid and augment biological experiments, hoping to help identify more accurate virus-host protein interaction maps. Moreover, computational methods can quickly adapt to predict how virus mutations change protein interactions with the host proteins.
We propose DeepVHPPI, a novel deep learning framework combining a self-attention-based transformer architecture and a transfer learning training strategy to predict interactions between human proteins and virus proteins that have novel sequence patterns. We show that our approach outperforms the state-of-the-art methods significantly in predicting Virus–Human protein interactions for SARS-CoV-2, H1N1, and Ebola. In addition, we demonstrate how our framework can be used to predict and interpret the interactions of mutated SARS-CoV-2 Spike protein sequences.
We make all of our data and code available on GitHub https://github.com/QData/DeepVHPPI.
Citations
@article {Lanchantin2020.12.14.422772,
author = {Lanchantin, Jack and Weingarten, Tom and Sekhon, Arshdeep and Miller, Clint and Qi, Yanjun},
title = {Transfer Learning for Predicting Virus-Host Protein Interactions for Novel Virus Sequences},
elocation-id = {2020.12.14.422772},
year = {2021},
doi = {10.1101/2020.12.14.422772},
publisher = {Cold Spring Harbor Laboratory},
abstract = {Viruses such as SARS-CoV-2 infect the human body by forming interactions between virus proteins and human proteins. However, experimental methods to find protein interactions are inadequate: large scale experiments are noisy, and small scale experiments are slow and expensive. Inspired by the recent successes of deep neural networks, we hypothesize that deep learning methods are well-positioned to aid and augment biological experiments, hoping to help identify more accurate virus-host protein interaction maps. Moreover, computational methods can quickly adapt to predict how virus mutations change protein interactions with the host proteins.We propose DeepVHPPI, a novel deep learning framework combining a self-attention-based transformer architecture and a transfer learning training strategy to predict interactions between human proteins and virus proteins that have novel sequence patterns. We show that our approach outperforms the state-of-the-art methods significantly in predicting Virus{\textendash}Human protein interactions for SARS-CoV-2, H1N1, and Ebola. In addition, we demonstrate how our framework can be used to predict and interpret the interactions of mutated SARS-CoV-2 Spike protein sequences.Availability We make all of our data and code available on GitHub https://github.com/QData/DeepVHPPI.ACM Reference Format Jack Lanchantin, Tom Weingarten, Arshdeep Sekhon, Clint Miller, and Yanjun Qi. 2021. Transfer Learning for Predicting Virus-Host Protein Interactions for Novel Virus Sequences. In Proceedings of ACM Conference (ACM-BCB). ACM, New York, NY, USA, 10 pages. https://doi.org/??Competing Interest StatementThe authors have declared no competing interest.},
URL = {https://www.biorxiv.org/content/early/2021/06/08/2020.12.14.422772},
eprint = {https://www.biorxiv.org/content/early/2021/06/08/2020.12.14.422772.full.pdf},
journal = {bioRxiv}
}
Having trouble with our tools? Please contact Jack and we’ll help you sort it out.
13 Apr 2021
On June 24th, 2021, I gave an invited talk at the Science Academy Machine Learning Summer School on “TextAttack: Generalizing Adversarial Examples to
Natural Language Processing”
Previous version of the tutorial: On April 14 2021, I gave an invited talk at the UVA Human and Machine Intelligence Seminar:
01 Mar 2021
Abstract
Multi-label image classification is the task of predicting a set of labels corresponding to objects, attributes or other entities present in an image. In this work we propose the Classification Transformer (C-Tran), a general framework for multi-label image classification that leverages Transformers to exploit the complex dependencies among visual features and labels. Our approach consists of a Transformer encoder trained to predict a set of target labels given an input set of masked labels, and visual features from a convolutional neural network. A key ingredient of our method is a label mask training objective that uses a ternary encoding scheme to represent the state of the labels as positive, negative, or unknown during training. Our model shows state-of-the-art performance on challenging datasets such as COCO and Visual Genome. Moreover, because our model explicitly represents the uncertainty of labels during training, it is more general by allowing us to produce improved results for images with partial or extra label annotations during inference. We demonstrate this additional capability in the COCO, Visual Genome, News500, and CUB image datasets.
Citations
@article{lanchantin2020general,
title={General Multi-label Image Classification with Transformers},
author={Jack Lanchantin and Tianlu Wang and Vicente Ordonez and Yanjun Qi},
year={2020},
eprint={2011.14027},
archivePrefix={arXiv, CVPR2021},
primaryClass={cs.CV}
}
Having trouble with our tools? Please contact Jack Lanchantin and we’ll help you sort it out.
11 Jan 2021
Title: Curriculum Labeling- Self-paced Pseudo-Labeling for Semi-Supervised Learning”
at the Thirty-Fifth AAAI Conference on Artificial Intelligence (AAAI-21) (acceptance rate: 21%))
authors: Paola Cascante-Bonilla, Fuwen Tan, Yanjun Qi, Vicente Ordonez
Abstract
In this paper we revisit the idea of pseudo-labeling in the context of semi-supervised learning where a learning algorithm has access to a small set of labeled samples and a large set of unlabeled samples. Pseudo-labeling works by applying pseudo-labels to samples in the unlabeled set by using a model trained on the combination of the labeled samples and any previously pseudo-labeled samples, and iteratively repeating this process in a self-training cycle. Current methods seem to have abandoned this approach in favor of consistency regularization methods that train models under a combination of different styles of self-supervised losses on the unlabeled samples and standard supervised losses on the labeled samples. We empirically demonstrate that pseudo-labeling can in fact be competitive with the state-of-the-art, while being more resilient to out-of-distribution samples in the unlabeled set. We identify two key factors that allow pseudo-labeling to achieve such remarkable results (1) applying curriculum learning principles and (2) avoiding concept drift by restarting model parameters before each self-training cycle. We obtain 94.91% accuracy on CIFAR-10 using only 4,000 labeled samples, and 68.87% top-1 accuracy on Imagenet-ILSVRC using only 10% of the labeled samples. The code is available at following https URL
Citations
@misc{grigsby2020measuring,
title={Measuring Visual Generalization in Continuous Control from Pixels},
author={Jake Grigsby and Yanjun Qi},
year={2020},
eprint={2010.06740},
archivePrefix={arXiv},
primaryClass={cs.LG}
}
Having trouble with our tools? Please contact Jake and we’ll help you sort it out.
11 Dec 2020
Title: Measuring Visual Generalization in Continuous Control from Pixels
- authors: Jake Grigsby, Yanjun Qi
Abstract
Self-supervised learning and data augmentation have significantly reduced the performance gap between state and image-based reinforcement learning agents in continuous control tasks. However, it is still unclear whether current techniques can face a variety of visual conditions required by real-world environments. We propose a challenging benchmark that tests agents’ visual generalization by adding graphical variety to existing continuous control domains. Our empirical analysis shows that current methods struggle to generalize across a diverse set of visual changes, and we examine the specific factors of variation that make these tasks difficult. We find that data augmentation techniques outperform self-supervised learning approaches and that more significant image transformations provide better visual generalization \footnote{The benchmark and our augmented actor-critic implementation are open-sourced @ this https URL)
Citations
@misc{grigsby2020measuring,
title={Measuring Visual Generalization in Continuous Control from Pixels},
author={Jake Grigsby and Yanjun Qi},
year={2020},
eprint={2010.06740},
archivePrefix={arXiv},
primaryClass={cs.LG}
}
Having trouble with our tools? Please contact Jake and we’ll help you sort it out.
01 Oct 2020
Title: Searching for a Search Method: Benchmarking Search Algorithms for Generating NLP Adversarial Examples
-
Abstract: We study the behavior of several black-box search algorithms used for generating adversarial examples for natural language processing (NLP) tasks. We perform a fine-grained analysis of three elements relevant to search: search algorithm, search space, and search budget. When new search methods are proposed in past work, the attack search space is often modified alongside the search method. Without ablation studies benchmarking the search algorithm change with the search space held constant, an increase in attack success rate could from an improved search method or a less restrictive search space. Additionally, many previous studies fail to properly consider the search algorithms’ run-time cost, which is essential for downstream tasks like adversarial training. Our experiments provide a reproducible benchmark of search algorithms across a variety of search spaces and query budgets to guide future research in adversarial NLP. Based on our experiments, we recommend greedy attacks with word importance ranking when under a time constraint or attacking long inputs, and either beam search or particle swarm optimization otherwise.
-
Citations:
@misc{yoo2020searching,
title={Searching for a Search Method: Benchmarking Search Algorithms for Generating NLP Adversarial Examples},
author={Jin Yong Yoo and John X. Morris and Eli Lifland and Yanjun Qi},
year={2020},
eprint={2009.06368},
archivePrefix={arXiv},
primaryClass={cs.CL}
}
Benchmarking Attack Recipes
-
As we emphasized in the above paper, we don’t recommend to directly compare Attack Recipes out of the box.
-
This is due to that attack recipes in the recent literature used different ways or thresholds in setting up their constraints. Without the constraint space held constant, an increase in attack success rate could come from an improved search or transformation method or a less restrictive search space.
01 Sep 2020
Title: Reevaluating Adversarial Examples in Natural Language
-
Paper EMNLP Findings
-
Abstract: State-of-the-art attacks on NLP models lack a shared definition of a what constitutes a successful attack. We distill ideas from past work into a unified framework: a successful natural language adversarial example is a perturbation that fools the model and follows some linguistic constraints. We then analyze the outputs of two state-of-the-art synonym substitution attacks. We find that their perturbations often do not preserve semantics, and 38% introduce grammatical errors. Human surveys reveal that to successfully preserve semantics, we need to significantly increase the minimum cosine similarities between the embeddings of swapped words and between the sentence encodings of original and perturbed sentences.With constraints adjusted to better preserve semantics and grammaticality, the attack success rate drops by over 70 percentage points.
- Citations
@misc{morris2020reevaluating,
title={Reevaluating Adversarial Examples in Natural Language},
author={John X. Morris and Eli Lifland and Jack Lanchantin and Yangfeng Ji and Yanjun Qi},
year={2020},
eprint={2004.14174},
archivePrefix={arXiv},
primaryClass={cs.CL}
}
Some of our evaluation results on quality of two SOTA attack recipes
- As we have emphasized in this paper, we recommend researchers and users to be EXTREMELY mindful on the quality of generated adversarial examples in natural language
- We recommend the field to use human-evaluation derived thresholds for setting up constraints
Some of our evaluation results on how to set constraints to evaluate NLP model’s adversarial robustness
12 May 2020
Title: TextAttack: A Framework for Adversarial Attacks in Natural Language Processing
Abstract
TextAttack is a library for generating natural language adversarial examples to fool natural language processing (NLP) models. TextAttack builds attacks from four components: a search method, goal function, transformation, and a set of constraints. Researchers can use these components to easily assemble new attacks. Individual components can be isolated and compared for easier ablation studies. TextAttack currently supports attacks on models trained for text classification and entailment across a variety of datasets. Additionally, TextAttack’s modular design makes it easily extensible to new NLP tasks, models, and attack strategies. TextAttack code and tutorials are available at this https URL.
It is a Python framework for adversarial attacks, data augmentation, and model training in NLP.

Citations
@misc{morris2020textattack,
title={TextAttack: A Framework for Adversarial Attacks in Natural Language Processing},
author={John X. Morris and Eli Lifland and Jin Yong Yoo and Yanjun Qi},
year={2020},
eprint={2005.05909},
archivePrefix={arXiv},
primaryClass={cs.CL}
}
Having trouble with our tools? Please contact Dr.Qi and we’ll help you sort it out.
01 May 2020
Title: Graph Convolutional Networks for Epigenetic State Prediction Using Both Sequence and 3D Genome Data
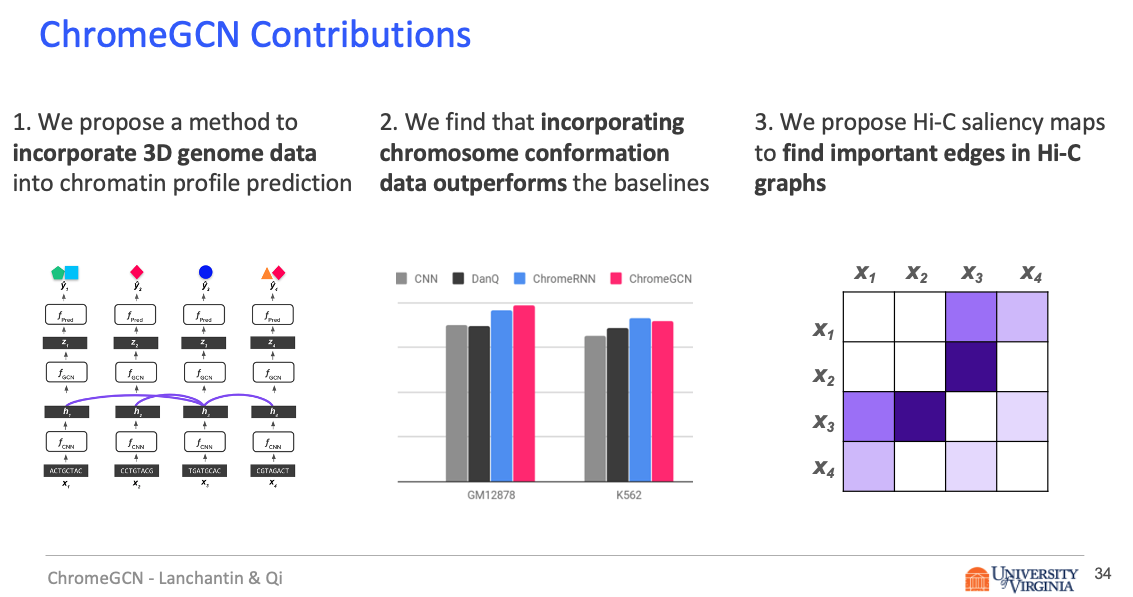
Abstract
Motivation
Predictive models of DNA chromatin profile (i.e. epigenetic state), such as transcription factor binding, are essential for understanding regulatory processes and developing gene therapies. It is known that the 3D genome, or spatial structure of DNA, is highly influential in the chromatin profile. Deep neural networks have achieved state of the art performance on chromatin profile prediction by using short windows of DNA sequences independently. These methods, however, ignore the long-range dependencies when predicting the chromatin profiles because modeling the 3D genome is challenging.
Results
In this work, we introduce ChromeGCN, a graph convolutional network for chromatin profile prediction by fusing both local sequence and long-range 3D genome information. By incorporating the 3D genome, we relax the independent and identically distributed assumption of local windows for a better representation of DNA. ChromeGCN explicitly incorporates known long-range interactions into the modeling, allowing us to identify and interpret those important long-range dependencies in influencing chromatin profiles. We show experimentally that by fusing sequential and 3D genome data using ChromeGCN, we get a significant improvement over the state-of-the-art deep learning methods as indicated by three metrics. Importantly, we show that ChromeGCN is particularly useful for identifying epigenetic effects in those DNA windows that have a high degree of interactions with other DNA windows.
Citations
@article{10.1093/bioinformatics/btaa793,
author = {Lanchantin, Jack and Qi, Yanjun},
title = "{Graph convolutional networks for epigenetic state prediction using both sequence and 3D genome data}",
journal = {Bioinformatics},
volume = {36},
number = {Supplement_2},
pages = {i659-i667},
year = {2020},
month = {12},
issn = {1367-4803},
doi = {10.1093/bioinformatics/btaa793},
url = {https://doi.org/10.1093/bioinformatics/btaa793},
eprint = {https://academic.oup.com/bioinformatics/article-pdf/36/Supplement\_2/i659/35336695/btaa793.pdf},
}
Having trouble with our tools? Please contact Jack and we’ll help you sort it out.
01 May 2020
Title: FastSK: Fast Sequence Analysis with Gapped String Kernels
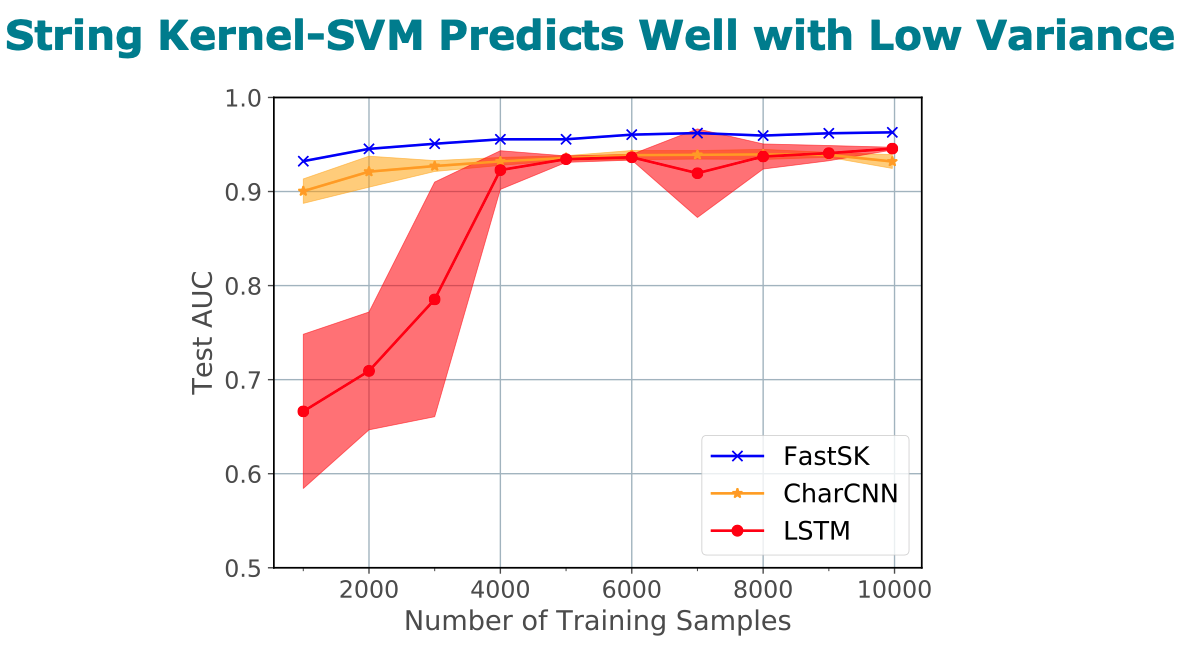

Abstract
Gapped k-mer kernels with Support Vector Machines (gkm-SVMs) have achieved strong predictive performance on regulatory DNA sequences
on modestly-sized training sets. However, existing gkm-SVM algorithms
suffer from the slow kernel computation time, as they depend
exponentially on the sub-sequence feature-length, number of mismatch
positions, and the task’s alphabet size.
In this work, we introduce a fast and scalable algorithm for
calculating gapped k-mer string kernels. Our method, named FastSK,
uses a simplified kernel formulation that decomposes the kernel
calculation into a set of independent counting operations over the
possible mismatch positions. This simplified decomposition allows us
to devise a fast Monte Carlo approximation that rapidly converges.
FastSK can scale to much greater feature lengths, allows us to
consider more mismatches, and is performant on a variety of sequence
analysis tasks. On 10 DNA transcription factor binding site (TFBS)
prediction datasets, FastSK consistently matches or outperforms the
state-of-the-art gkmSVM-2.0 algorithms in AUC, while achieving
average speedups in kernel computation of 100 times and speedups of
800 times for large feature lengths. We further show that FastSK
outperforms character-level recurrent and convolutional neural
networks across all 10 TFBS tasks. We then extend FastSK to 7
English medical named entity recognition datasets and 10 protein
remote homology detection datasets. FastSK consistently matches or
outperforms these baselines.
Our algorithm is available as a Python package and as C++ source code.
(Available for download at https://github.com/Qdata/FastSK/.
Install with the command make or pip install)
Citations
@article{10.1093/bioinformatics/btaa817,
author = {Blakely, Derrick and Collins, Eamon and Singh, Ritambhara and Norton, Andrew and Lanchantin, Jack and Qi, Yanjun},
title = "{FastSK: fast sequence analysis with gapped string kernels}",
journal = {Bioinformatics},
volume = {36},
number = {Supplement_2},
pages = {i857-i865},
year = {2020},
month = {12},
issn = {1367-4803},
doi = {10.1093/bioinformatics/btaa817},
url = {https://doi.org/10.1093/bioinformatics/btaa817},
eprint = {https://academic.oup.com/bioinformatics/article-pdf/36/Supplement\_2/i857/35337038/btaa817.pdf},
}
Having trouble with our tools? Please contact Yanjun Qi and we’ll help you sort it out.
30 Jul 2019
Here is the slide of my tutorial talk at UCLA computational genomics summer school 2019.
Slides: PDF
Thanks for reading!
18 Jul 2019
JointNets R in CRAN : URL
Github Site: URL
Talk slide by Zhaoyang about the jointnet implementations:
Demo GUI Run:
Demo Visualization of a few learned networks:
- DIFFEE on one gene expression dataset about breast cancer
- JEEK on one simulated data about samples from multiple contexts and nodes with extra spatial information
- SIMULE on one word based text dataset including multiple categories
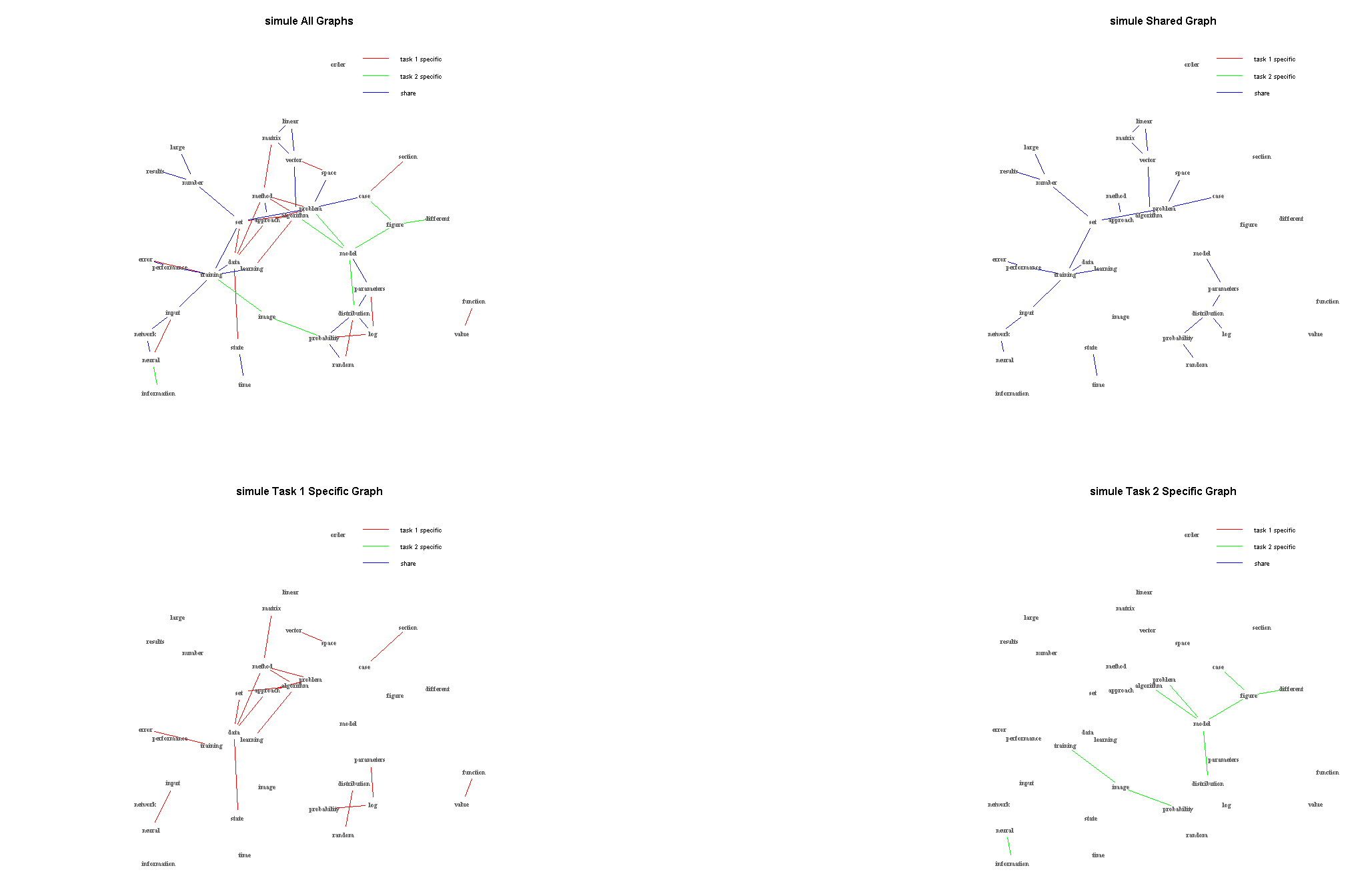
- SIMULE on one multi-context Brain fMRI dataset

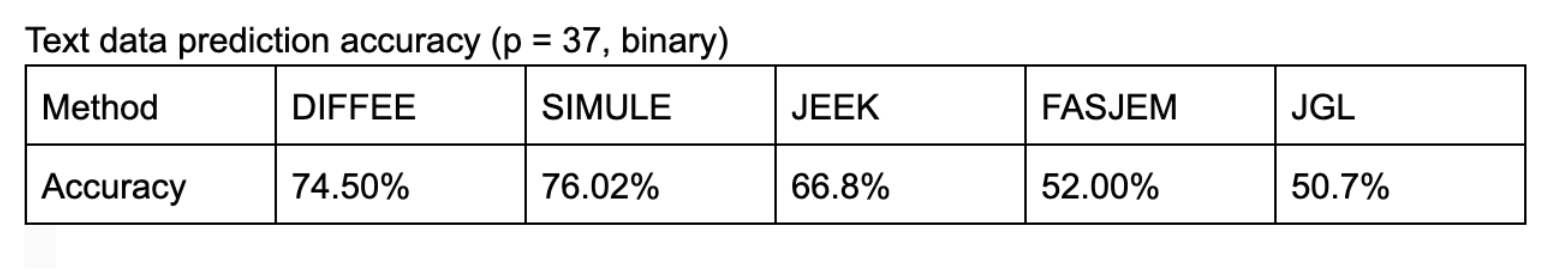
- Demo downstream task using learned graphs for classification, e.g., on a two class text dataset, we get
- With Zoom In/Out function
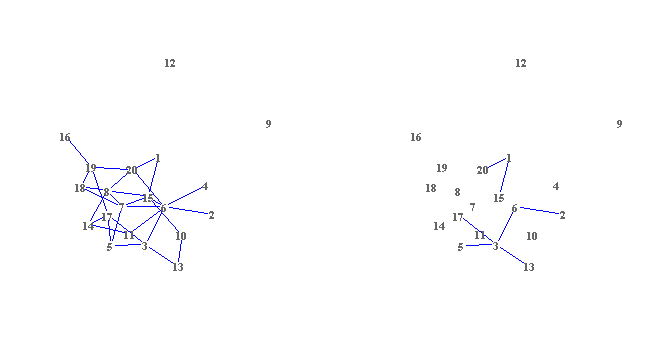
- With Multiple window design, legend, title coloring schemes
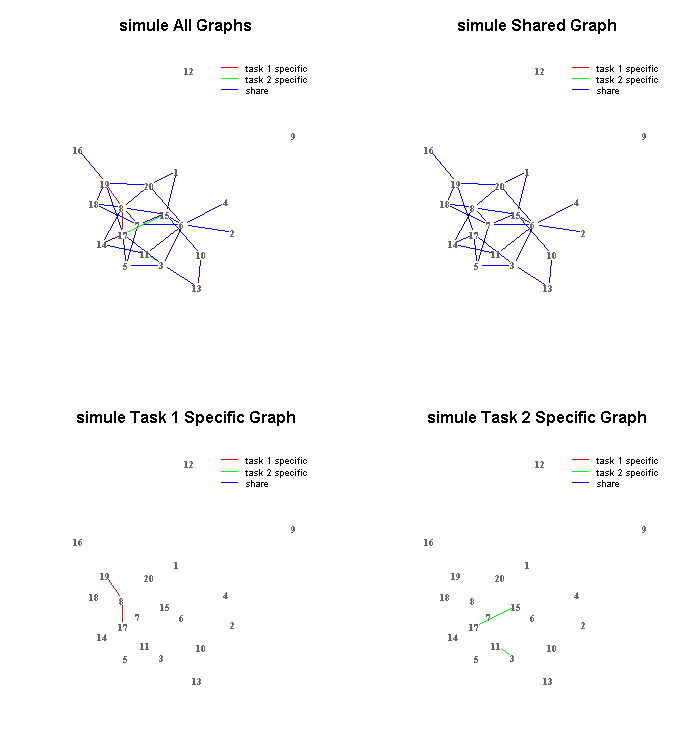
Flow charts of the code design (functional and module level) in jointnets package
Citations
@conference{wang2018jeek,
Author = {Wang, Beilun and Sekhon, Arshdeep and Qi, Yanjun},
Booktitle = {Proceedings of The 35th International Conference on Machine Learning (ICML)},
Title = {A Fast and Scalable Joint Estimator for Integrating Additional Knowledge in Learning Multiple Related Sparse Gaussian Graphical Models},
Year = {2018}}
}
Having trouble with our tools? Please contact Arsh and we’ll help you sort it out.
18 Jun 2019
Abstract
We focus on integrating different types of extra knowledge (other than the observed samples) for estimating the sparse structure change between two p-dimensional Gaussian Graphical Models (i.e. differential GGMs). Previous differential GGM estimators either fail to include additional knowledge or cannot scale up to a high-dimensional (large p) situation. This paper proposes a novel method KDiffNet that incorporates Additional Knowledge in identifying Differential Networks via an Elementary Estimator. We design a novel hybrid norm as a superposition of two structured norms guided by the extra edge information and the additional node group knowledge. KDiffNet is solved through a fast parallel proximal algorithm, enabling it to work in large-scale settings. KDiffNet can incorporate various combinations of existing knowledge without re-designing the optimization. Through rigorous statistical analysis we show that, while considering more evidence, KDiffNet achieves the same convergence rate as the state-of-the-art. Empirically on multiple synthetic datasets and one real-world fMRI brain data, KDiffNet significantly outperforms the cutting edge baselines concerning the prediction performance, while achieving the same level of time cost or less.
Citations
@conference{arsh19kdiffNet,
Author = {Sekhon, Arshdeep and Wang, Beilun and Qi, Yanjun},
Title = {Adding Extra Knowledge in Scalable Learning of
Sparse Differential Gaussian Graphical Models},
Year = {2019}}
}
Having trouble with our tools? Please contact Arsh and we’ll help you sort it out.
15 Apr 2019
Ph.D. Dissertation Defense by Weilin Xu
Title: Improving Robustness of Machine Learning Models using Domain Knowledge
- 11 am, Monday, April 15, 2019, at Rice 504.
Committee Members:
Vicente Ordonezan (Committee Chair),
David Evans, Advisor
Yanjun Qi, Advisor
Homa Alemzadeh
Patrick McDaniel
Abstract
Although machine learning techniques have achieved great success in many areas, such as computer vision, natural language processing, and computer security, recent studies have shown that they are not robust under attack. A motivated adversary is often able to craft input samples that force a machine learning model to produce incorrect predictions, even if the target model achieves high accuracy on normal test inputs. This raises great concern when machine learning models are deployed for security-sensitive tasks.
This dissertation aims to improve the robustness of machine learning models by exploiting domain knowledge. While domain knowledge has often been neglected due to the power of automatic representation learning in the deep learning era, we find that domain knowledge goes beyond a given dataset of a task and helps to (1) uncover weaknesses of machine learning models, (2) detect adversarial examples and (3) improve the robustness of machine learning models.
First, we design an evolutionary algorithm-based framework, \emph{Genetic Evasion}, to find evasive samples. We embed domain knowledge into the mutation operator and the fitness function of the framework and achieve 100% success rate in evading two state-of-the-art PDF malware classifiers. Unlike previous methods, our technique uses genetic programming to directly generate evasive samples in the problem space instead of the feature space, making it a practical attack that breaks the trust of black-box machine learning models in a security application.
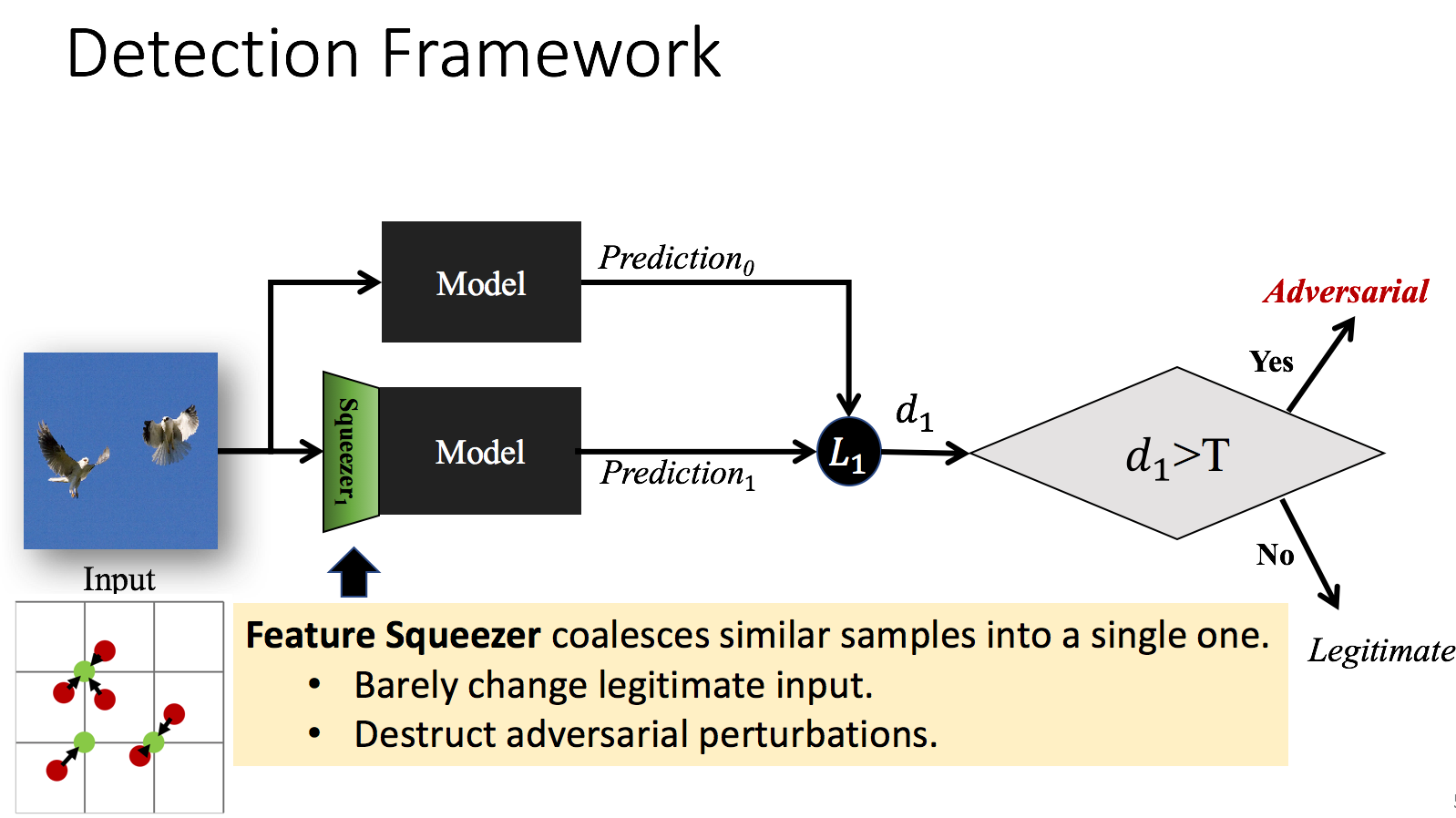
Second, we design an ensemble framework, \emph{Feature Squeezing}, to detect adversarial examples against deep neural network models using simple pre-processing. We employ domain knowledge on signal processing that natural signals are often redundant for many perception tasks. Therefore, we can squeeze the input features to reduce adversaries’ search space while preserving the accuracy on normal inputs.
We use various squeezers to pre-process an input example before it is fed into a model. The difference between those predictions is often small for normal inputs due to redundancy, while the difference can be large for adversarial examples. We demonstrate that \emph{Feature Squeezing} is empirically effective and inexpensive in detecting adversarial examples for image classification tasks generated by many algorithms.
Third, we incorporate simple pre-processing with certifiable robust training and formal verification to train provably-robust models. We formally analyze the impact of pre-processing on adversarial strength and derive novel methods to improve model robustness. Our approach produces accurate models with verified state-of-the-art robustness and advances the state-of-the-art of certifiable robust training methods.
We demonstrate that domain knowledge helps us understand and improve the robustness of machine learning models. Our results have motivated several subsequent works, and we hope this dissertation will be a step towards implementing robust models under attack.
03 Apr 2019
On April 23 2019, I gave an invited talk at the ARO Invitational Workshop on Foundations of Autonomous Adaptive Cyber Systems
01 Mar 2019
Title: Neural Message Passing for Multi-Label Classification
Abstract
Multi-label classification (MLC) is the task of assigning a set of target labels for a given sample. Modeling the combinatorial label interactions in MLC has been a long-haul challenge. Recurrent neural network (RNN) based encoder-decoder models have shown state-of-the-art performance for solving MLC. However, the sequential nature of modeling label dependencies through an RNN limits its ability in parallel computation, predicting dense labels, and providing interpretable results. In this paper, we propose Message Passing Encoder-Decoder (MPED) Networks, aiming to provide fast, accurate, and interpretable MLC. MPED networks model the joint prediction of labels by replacing all RNNs in the encoder-decoder architecture with message passing mechanisms and dispense with autoregressive inference entirely. The proposed models are simple, fast, accurate, interpretable, and structure-agnostic (can be used on known or unknown structured data). Experiments on seven real-world MLC datasets show the proposed models outperform autoregressive RNN models across five different metrics with a significant speedup during training and testing time.
Citations
@article{lanchantin2018neural,
title={Neural Message Passing for Multi-Label Classification},
author={Lanchantin, Jack and Sekhon, Arshdeep and Qi, Yanjun},
year={2018}
}
Having trouble with our tools? Please contact Jack Lanchantin and we’ll help you sort it out.
21 Dec 2018
On December 21 @ 12noon, I gave a distinguished webinar talk in the Fall 2018 webinar series of the Institute for Information Infrastructure Protection (I3P) (@ the George Washington University and SRI International).
Webinar Recording @ URL
10 Oct 2018
I gave a tutorial talk at
UVA-CPHG Seminar Series 2018.
Title: Making Deep Learning Understandable for Analyzing Sequential Data about Gene Regulation
Slides @:PDF
Thanks for reading!
07 Sep 2018
Paper:
Abstract:
Computational methods that predict differential gene expression from histone modification signals are highly desirable for understanding how histone modifications control the functional heterogeneity of cells through influencing differential gene regulation. Recent studies either failed to capture combinatorial effects on differential prediction or primarily only focused on cell type-specific analysis. In this paper, we develop a novel attention-based deep learning architecture, DeepDiff, that provides a unified and end-to-end solution to model and to interpret how dependencies among histone modifications control the differential patterns of gene regulation. DeepDiff uses a hierarchy of multiple Long short-term memory (LSTM) modules to encode the spatial structure of input signals and to model how various histone modifications cooperate automatically. We introduce and train two levels of attention jointly with the target prediction, enabling DeepDiff to attend differentially to relevant modifications and to locate important genome positions for each modification. Additionally, DeepDiff introduces a novel deep-learning based multi-task formulation to use the cell-type-specific gene expression predictions as auxiliary tasks, encouraging richer feature embeddings in our primary task of differential expression prediction. Using data from Roadmap Epigenomics Project (REMC) for ten different pairs of cell types, we show that DeepDiff significantly outperforms the state-of-the-art baselines for differential gene expression prediction. The learned attention weights are validated by observations from previous studies about how epigenetic mechanisms connect to differential gene expression. Codes and results are available at deepchrome.net
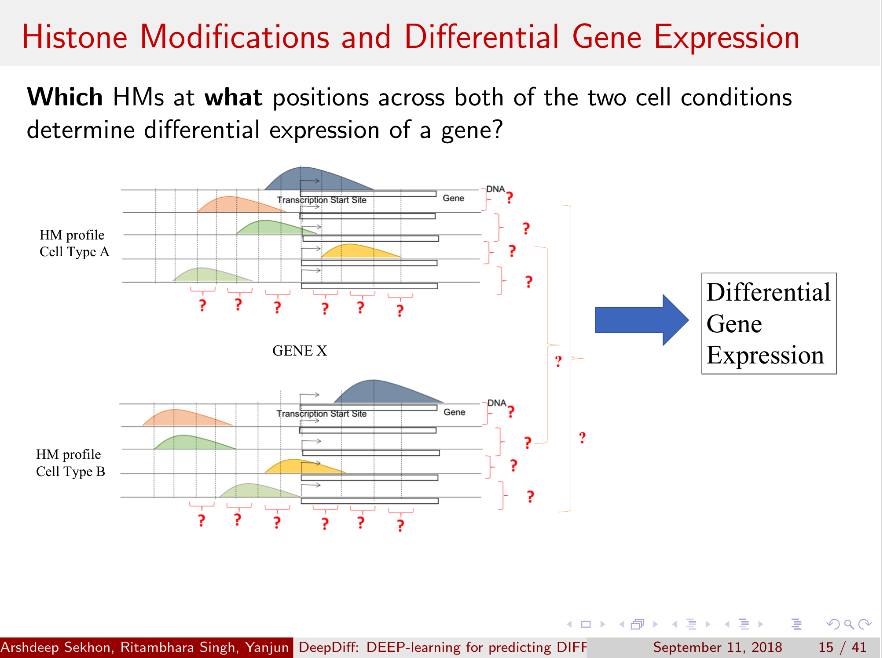
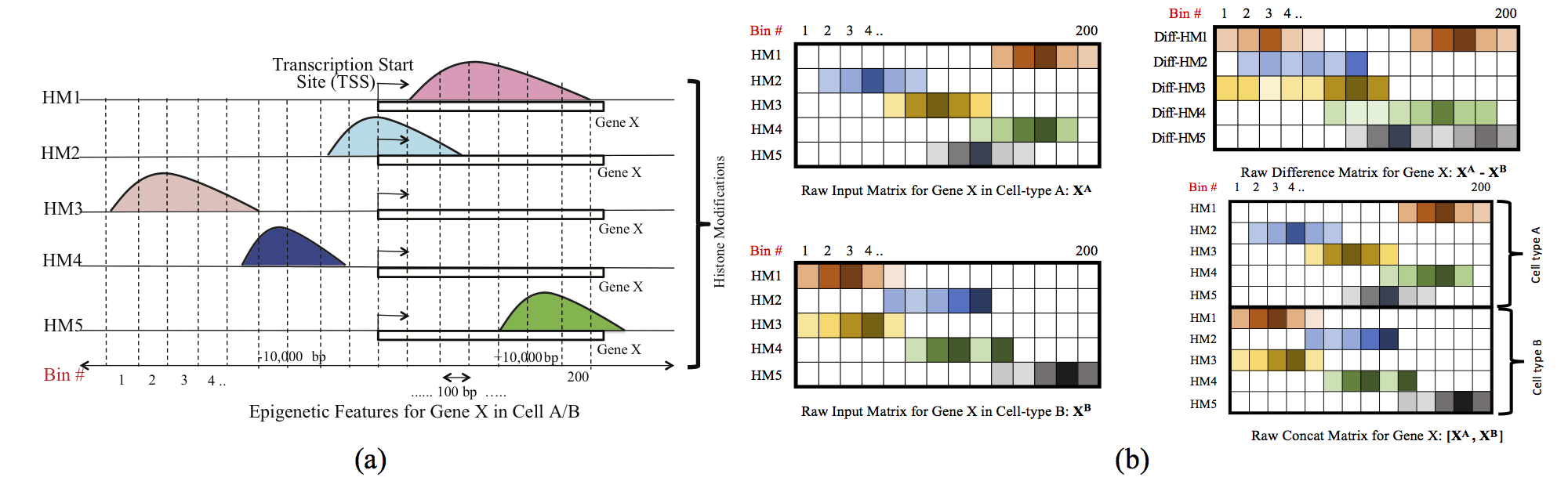
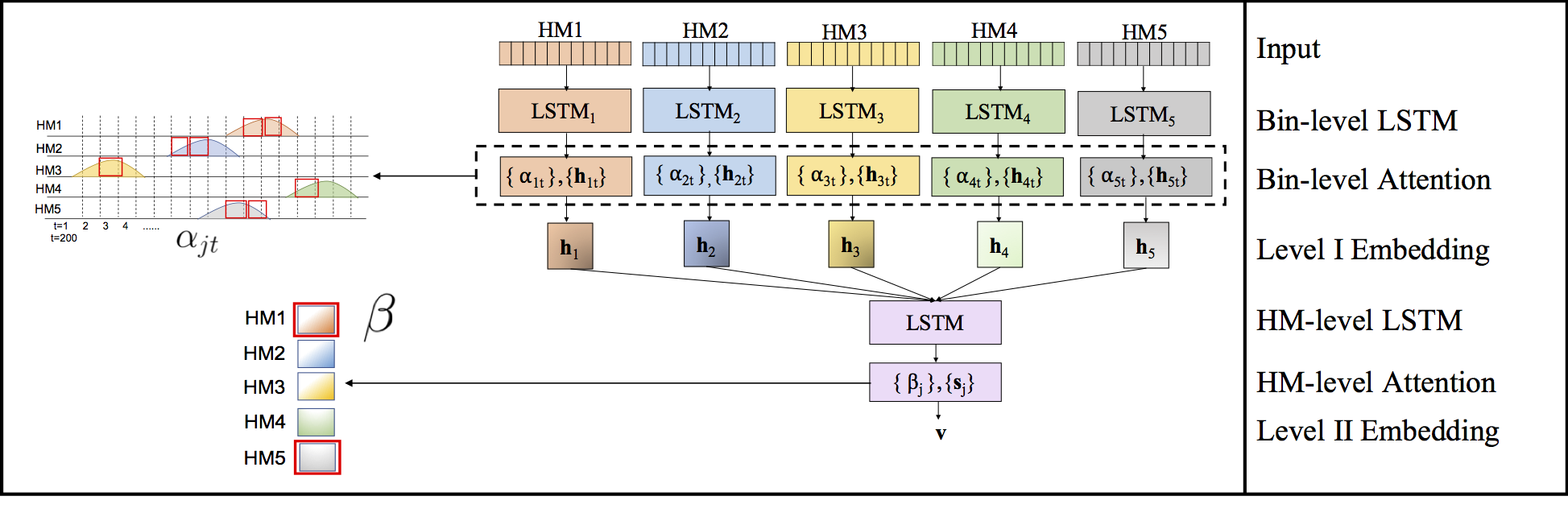
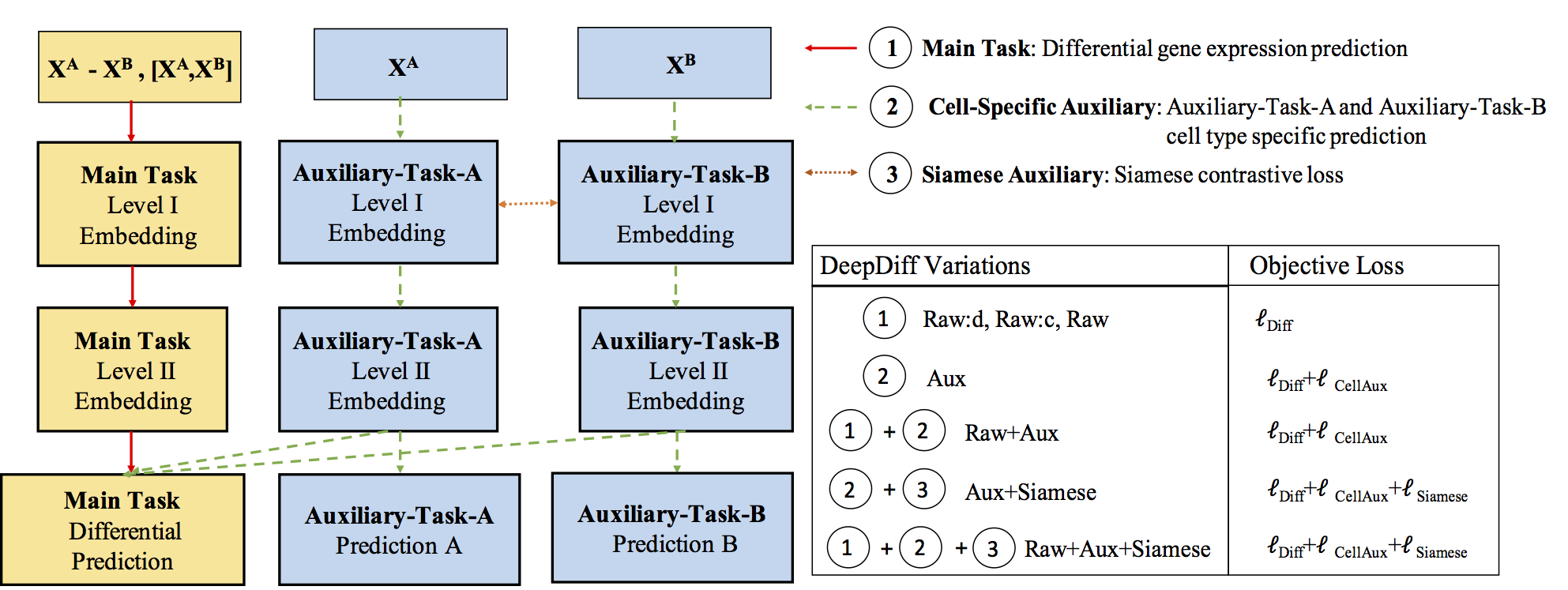
Citations
@article{ArDeepDiff18,
author = {Sekhon, Arshdeep and Singh, Ritambhara and Qi, Yanjun},
title = {DeepDiff: DEEP-learning for predicting DIFFerential gene expression from histone modifications},
journal = {Bioinformatics},
volume = {34},
number = {17},
pages = {i891-i900},
year = {2018},
doi = {10.1093/bioinformatics/bty612},
URL = {http://dx.doi.org/10.1093/bioinformatics/bty612},
eprint = {/oup/backfile/content_public/journal/bioinformatics/34/17/10.1093_bioinformatics_bty612/2/bty612.pdf}
}
Having trouble with our tools? Please contact Arsh and we’ll help you sort it out.
29 Aug 2018
Here are the slides of tutorial talk I gave at ACM-BCB 2018.
Title: Making Deep Learning Understandable for Analyzing Sequential Data about Gene Regulation
Part I Slides: PDF
Part II Slides:PDF
Thanks for reading!
19 Aug 2018
So far, we have released the following Tutorials:
Have questions or suggestions? Feel free to ask me on Twitter or email me.
Thanks for reading!
20 Jul 2018
PhD Defense Presentation by Beilun Wang
- Friday, July 20, 2018 at 9:00 am in Rice 242
- Committee Members: Mohammad Mahmoody (Chair), Yanjun Qi (Advisor), Farzad Farnoud, Xiaojin (Jerry) Zhu (University of Wisconsin–Madison), and Tingting Zhang (Statistics)
Title: Fast and Scalable Joint Estimators for Learning Sparse Gaussian Graphical Models from Heterogeneous Data with Additional Knowledge
- Abstract
Understanding and quantifying variable graphs from heterogeneous samples is a fundamental and urgent analysis task thanks to the data explosion in many scientific domains. Such variable graphs can significantly improve network-driven studies like understanding genetic or neural pathways or providing valuable tools for the discovery of therapeutic targets or diagnostic markers. One typical approach is to jointly estimate K different but related conditional dependency graphs through a multi-task formulation of the sparse Gaussian Graphical Model (multi-sGGM). Most current studies of multi-sGGMs, however, involve expensive and difficult non-smooth optimizations, making them difficult to scale up to many dimensions (large p) or with many contexts (large K).
In this dissertation, we aim to fill the gap and have designed a category of novel estimators that can achieve fast and scalable joint structure estimation of multiple sGGMs.
Three crucial tasks exist when learning multi-sGGMs from heterogeneous samples: (1) to enforce graph relatedness through structural norms, (2) to estimate the change of variable dependencies directly, and (3) to incorporate existing knowledge of the variable nodes or about relationships among nodes. Targeting each, our work introduces fast and parallelizable estimators that largely improves the computational efficiency of the state-of-the-art. We have conducted rigorous statistical analysis and verified that surprisingly the proposed estimators achieve the same statistical convergence rates as the state-of-art solutions that are much harder to compute. Empirically, our estimators outperform the speed of the cutting edge significantly while achieving the same or better prediction accuracy. We have implemented all proposed estimators into publicly accessible tools in the R-CRAN repository. This suite of toolboxes can help users effectively translate aggregated data into knowledge that take the form of graphs.
12 May 2018
Paper: Most updated version at HERE | Previous version: @Arxiv |
TalkSlide: URL
R package: URL
GitRepo for R package: URL
install.packages("jeek")
library(jeek)
demo(jeek)
Abstract
We consider the problem of including additional knowledge in estimating sparse Gaussian graphical models (sGGMs) from aggregated samples, arising often in bioinformatics and neuroimaging applications. Previous joint sGGM estimators either fail to use existing knowledge or cannot scale-up to many tasks (large $K$) under a high-dimensional (large $p$) situation. In this paper, we propose a novel \underline{J}oint \underline{E}lementary \underline{E}stimator incorporating additional \underline{K}nowledge (JEEK) to infer multiple related sparse Gaussian Graphical models from large-scale heterogeneous data. Using domain knowledge as weights, we design a novel hybrid norm as the minimization objective to enforce the superposition of two weighted sparsity constraints, one on the shared interactions and the other on the task-specific structural patterns. This enables JEEK to elegantly consider various forms of existing knowledge based on the domain at hand and avoid the need to design knowledge-specific optimization. JEEK is solved through a fast and entry-wise parallelizable solution that largely improves the computational efficiency of the state-of-the-art $O(p^5K^4)$ to $O(p^2K^4)$. We conduct a rigorous statistical analysis showing that JEEK achieves the same convergence rate $O(\log(Kp)/n_{tot})$ as the state-of-the-art estimators that are much harder to compute.
Empirically, on multiple synthetic datasets and two real-world data, JEEK outperforms the speed of the state-of-arts significantly while achieving the same level of prediction accuracy.
About Adding Additional Knowledge
One significant caveat of state-of-the-art joint sGGM estimators is the fact that little attention has been paid to incorporating existing knowledge of the nodes or knowledge of the relationships among nodes in the models.
In addition to the samples themselves, additional information is widely available in real-world applications. In fact, incorporating the knowledge is of great scientific interest. A prime example is when estimating the functional brain connectivity networks among brain regions based on fMRI samples, the spatial position of the regions are readily available. Neuroscientists have gathered considerable knowledge regarding the spatial and anatomical evidence underlying brain connectivity (e.g., short edges and certain anatomical regions are more likely to be connected \cite{watts1998collective}). Another important example is the problem of identifying gene-gene interactions from patients’ gene expression profiles across multiple cancer types. Learning the statistical dependencies among genes from such heterogeneous datasets can help to understand how such dependencies vary from normal to abnormal and help to discover contributing markers that influence or cause the diseases. Besides the patient samples, state-of-the-art bio-databases like HPRD \cite{prasad2009human} have collected a significant amount of information about direct physical interactions among corresponding proteins, regulatory gene pairs or signaling relationships collected from high-qualify bio-experiments.
Although being strong evidence of structural patterns we aim to discover, this type of information has rarely been considered in the joint sGGM formulation of such samples. This paper aims to fill this gap by adding additional knowledge most effectively into scalable and fast joint sGGM estimations.
The proposed JEEK estimator provides the flexibility of using ($K+1$) different weight matrices representing the extra knowledge. We try to showcase a few possible designs of the weight matrices, including (but not limited to):
- Spatial or anatomy knowledge about brain regions;
- Knowledge of known co-hub nodes or perturbed nodes;
- Known group information about nodes, such as genes belonging to the same biological pathway or cellular location;
- Using existing known edges as the knowledge, like the known protein interaction databases for discovering gene networks (a semi-supervised setting for such estimations).
We sincerely believe the scalability and flexibility provided by JEEK can make structure learning of joint sGGM feasible in many real-world tasks.
an example W for how to add known group sparity
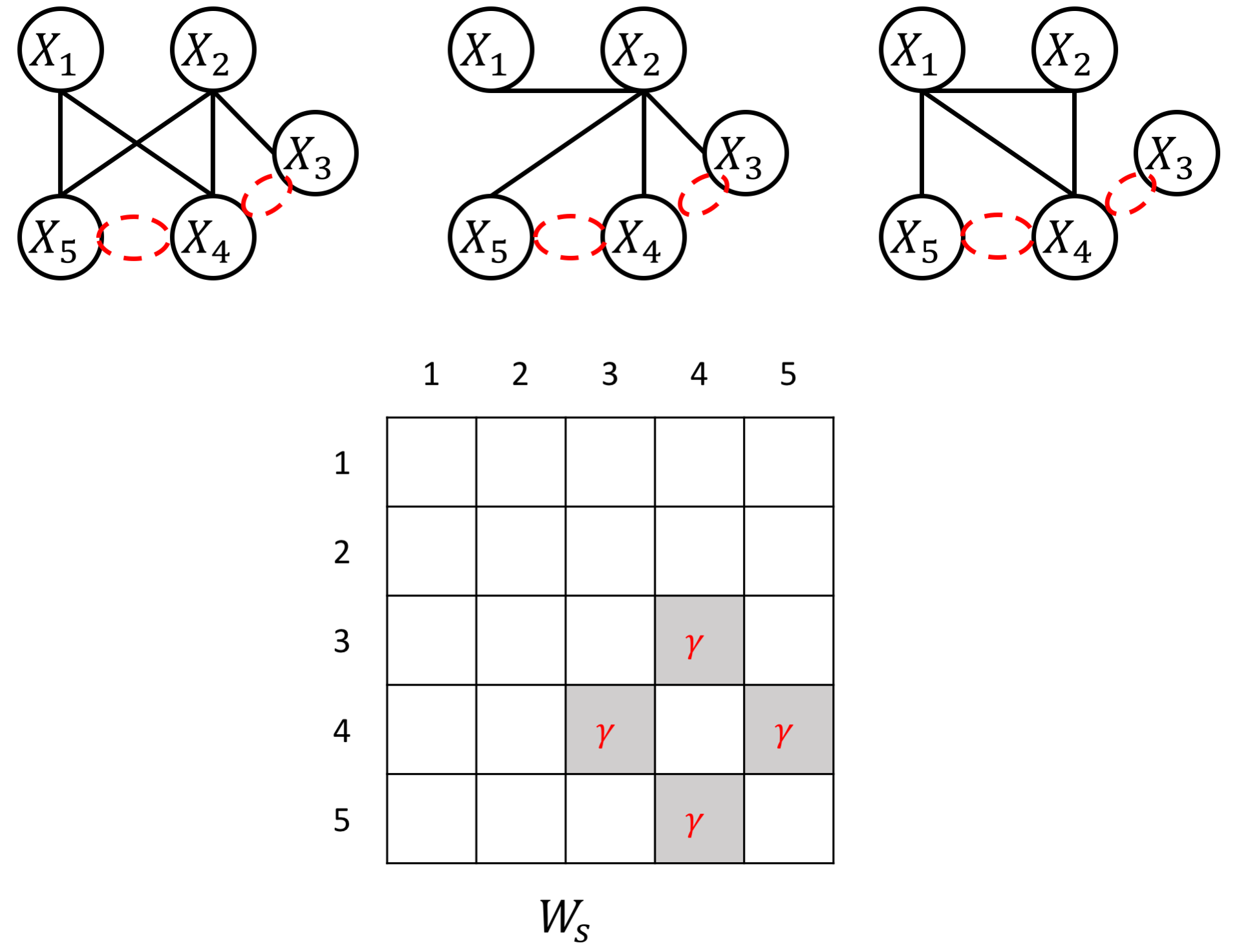
an example W for how to add known group interactions
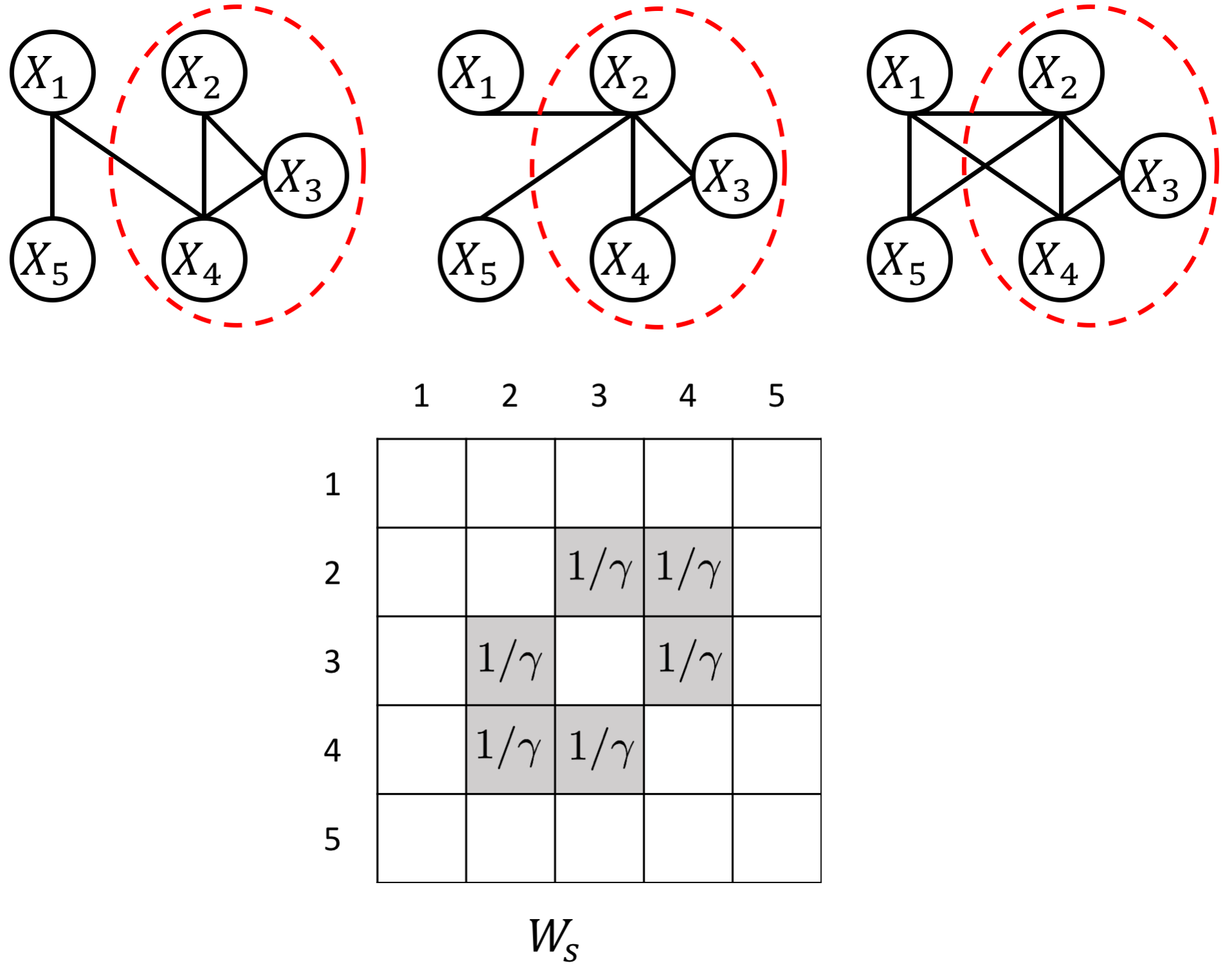
an example W for how to add known hub node
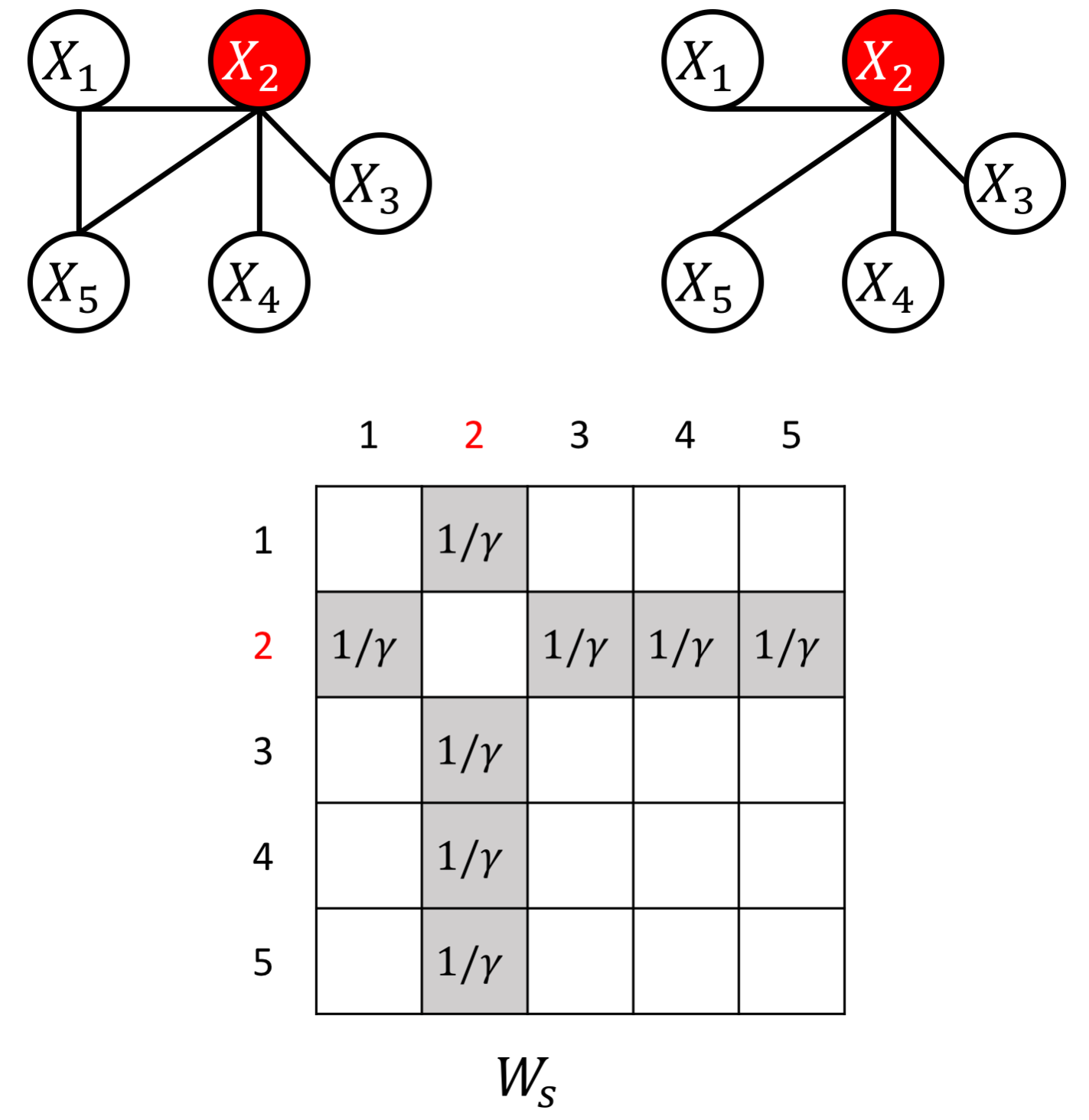
an example W for how to add known perturbed-hub node
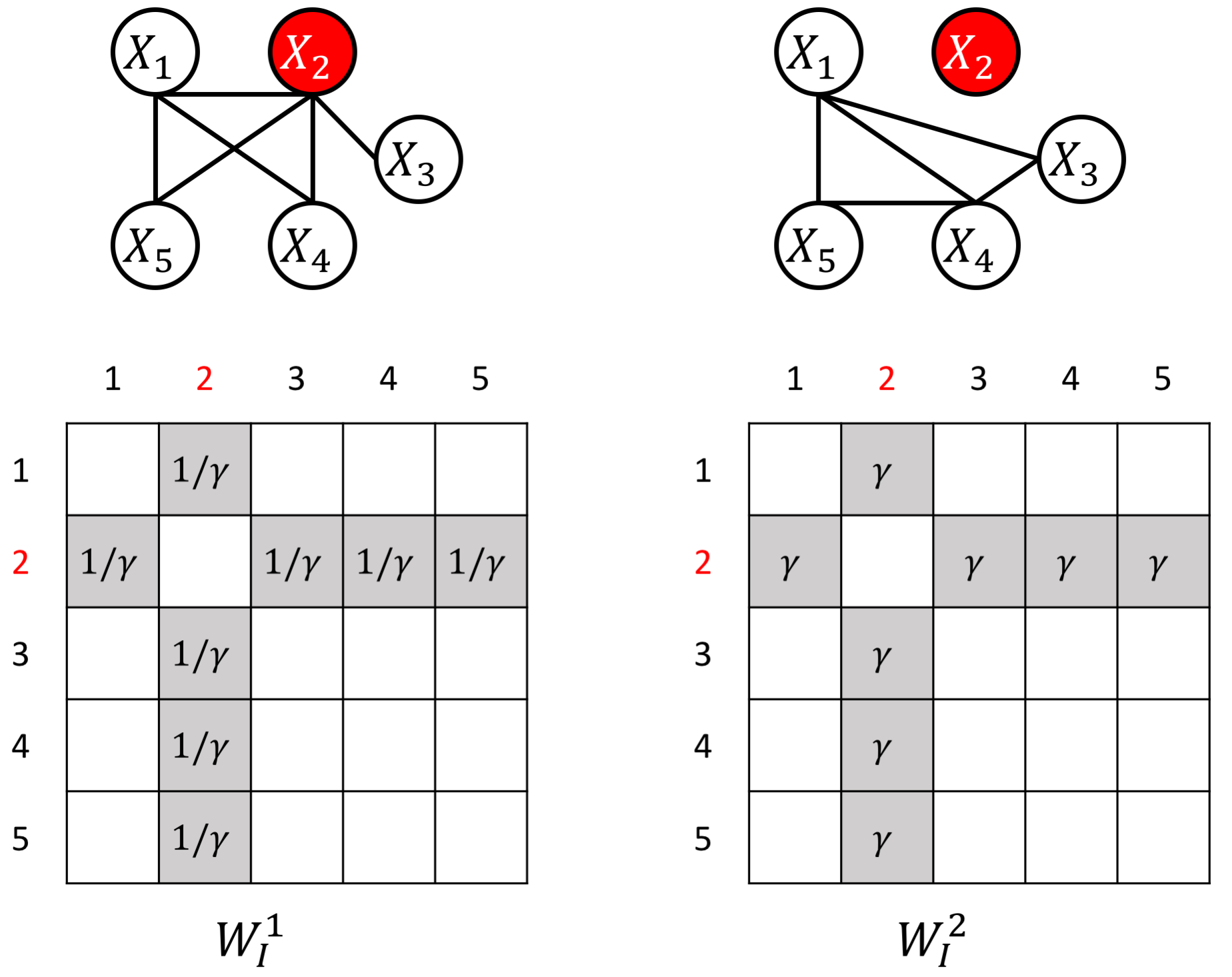
Citations
@conference{wang2018jeek,
Author = {Wang, Beilun and Sekhon, Arshdeep and Qi, Yanjun},
Booktitle = {Proceedings of The 35th International Conference on Machine Learning (ICML)},
Title = {A Fast and Scalable Joint Estimator for Integrating Additional Knowledge in Learning Multiple Related Sparse Gaussian Graphical Models},
Year = {2018}}
}
Having trouble with our tools? Please contact Beilun and we’ll help you sort it out.
09 Apr 2018
Ph.D. Dissertation Defense by Ritambhara Singh
- Monday, April 9, 2018 at 12:00PM in Rice 504.
Title: Fast and Interpretable Classification of Sequential Data in Biology
- Committee Members: Yanjun Qi (Advisor), Mary Lou Soffa (Chair), Gabriel Robins, Mazhar Adli (UVA Biochemistry and Molecular Genetics), Christina Leslie (Minor Representative- Memorial Sloan Kettering Cancer Center)
Abstract:
Biological sciences are rapidly becoming data intensive. Between 100 million to 2 billion human genomes are estimated to be sequenced by the year 2025, far exceeding the growth of big data domains like Astronomy, YouTube, and Twitter. Majority of these biological datasets are sequential in nature, representing the human genome as well as measurements of activity taking place around it. Analyzing this enormous repository of sequential data is both urgent and essential to understand genetic diseases and drug development. Data-driven approaches like machine learning have shown significant progress in analyzing the existing data. However, the state-of-the-art machine learning techniques face two hard challenges in this domain: (1) Interpretability of the predictions for better insights, and (2) Slow computation due to expanding search space of sequential patterns. In this dissertation, we aim to solve these two challenges by improving two popular machine learning models: Deep Neural Networks (DNNs) and String Kernel with Support Vector Machines (SK-SVM).
+[Challenge(1):] DNNs can handle large sequential datasets accurately and in an efficient manner. However, DNNs have widely been viewed as ‘black boxes’ due to the complex, multi-layer structure, making them hard to understand. We implement a unified DNN architecture to model and to interpret features in an end-to-end manner. The proposed design is not only accurate, but it also provides better interpretation than state-of-the-art feature visualization methods such as saliency maps.
+[Challenge (2):] SK-SVM methods achieve high accuracy and have theoretical guarantees with limited labeled training samples. However, current implementations run extremely slow when we increase the dictionary size or allow more mismatches. We present a novel algorithmic implementation for calculating Gapped k-mer string Kernel using Counting (GaKCo). This method is fast, scalable and naturally parallelizable. Empirically, GaKCo performs up to 100 times faster than the state-of-the-art SK-SVM method across multiple biological sequential datasets.
08 Mar 2018
Here are the slides of lecture talks I gave at UCLA CGWI and NLM-CBB seminar about our deep learning tools: DeepChrome, AttentiveChrome and DeepMotif.
Slides: PDF

Thanks for reading!
02 Mar 2018
Abstract
Although deep neural networks (DNNs) have achieved great success in many computer vision tasks, recent studies have shown they are vulnerable to adversarial examples. Such examples, typically generated by adding small but purposeful distortions, can frequently fool DNN models. Previous studies to defend against adversarial examples mostly focused on refining the DNN models. They have either shown limited success or suffer from the expensive computation. We propose a new strategy, \emph{feature squeezing}, that can be used to harden DNN models by detecting adversarial examples. Feature squeezing reduces the search space available to an adversary by coalescing samples that correspond to many different feature vectors in the original space into a single sample. By comparing a DNN model’s prediction on the original input with that on the squeezed input, feature squeezing detects adversarial examples with high accuracy and few false positives. This paper explores two instances of feature squeezing: reducing the color bit depth of each pixel and smoothing using a spatial filter. These strategies are straightforward, inexpensive, and complementary to defensive methods that operate on the underlying model, such as adversarial training.
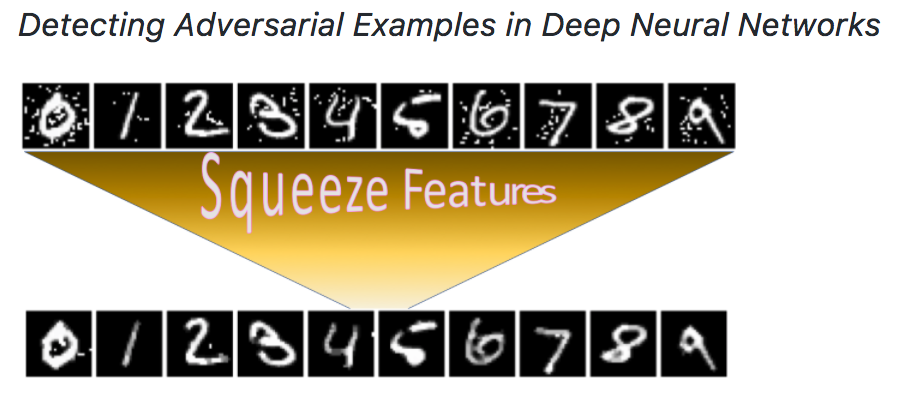
Citations
@inproceedings{Xu0Q18,
author = {Weilin Xu and
David Evans and
Yanjun Qi},
title = {Feature Squeezing: Detecting Adversarial Examples in Deep Neural Networks},
booktitle = {25th Annual Network and Distributed System Security Symposium, {NDSS}
2018, San Diego, California, USA, February 18-21, 2018},
year = {2018},
crossref = {DBLP:conf/ndss/2018},
url = {http://wp.internetsociety.org/ndss/wp-content/uploads/sites/25/2018/02/ndss2018\_03A-4\_Xu\_paper.pdf},
timestamp = {Thu, 09 Aug 2018 10:57:16 +0200},
biburl = {https://dblp.org/rec/bib/conf/ndss/Xu0Q18},
bibsource = {dblp computer science bibliography, https://dblp.org}
}
Having troubl with our tools? Please contact Weilin and we’ll help you sort it out.
12 Jan 2018
Title: Black-box Generation of Adversarial Text Sequences to Fool Deep Learning Classifiers
TalkSlide: URL
Published @ 2018 IEEE Security and Privacy Workshops (SPW),
co-located with the 39th IEEE Symposium on Security and Privacy.
Abstract
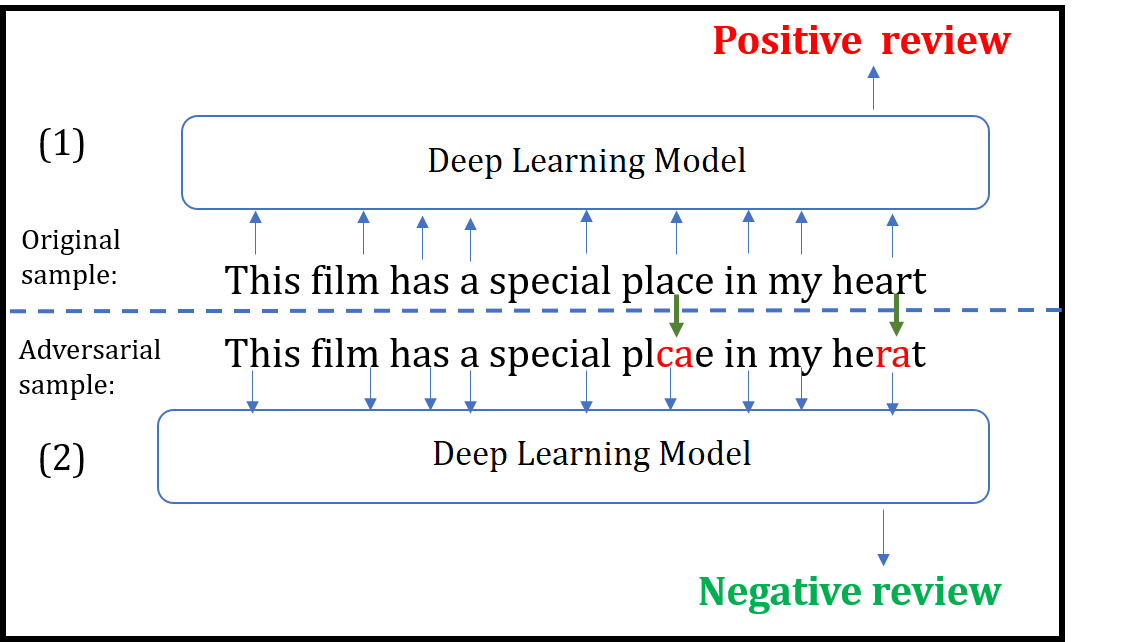
Although various techniques have been proposed to generate adversarial samples for white-box attacks on text, little attention has been paid to a black-box attack, which is a more realistic scenario. In this paper, we present a novel algorithm, DeepWordBug, to effectively generate small text perturbations in a black-box setting that forces a deep-learning classifier to misclassify a text input. We develop novel scoring strategies to find the most important words to modify such that the deep classifier makes a wrong prediction. Simple character-level transformations are applied to the highest-ranked words in order to minimize the edit distance of the perturbation. We evaluated DeepWordBug on two real-world text datasets: Enron spam emails and IMDB movie reviews. Our experimental results indicate that DeepWordBug can reduce the classification accuracy from 99% to around 40% on Enron data and from 87% to about 26% on IMDB. Also, our experimental results strongly demonstrate that the generated adversarial sequences from a deep-learning model can similarly evade other deep models.
We build an interactive extension to visualize DeepWordbug:
- Interactive Live Demo @ ULR
Citations
@INPROCEEDINGS{JiDeepWordBug18,
author={J. Gao and J. Lanchantin and M. L. Soffa and Y. Qi},
booktitle={2018 IEEE Security and Privacy Workshops (SPW)},
title={Black-Box Generation of Adversarial Text Sequences to Evade Deep Learning Classifiers},
year={2018},
pages={50-56},
keywords={learning (artificial intelligence);pattern classification;program debugging;text analysis;deep learning classifiers;character-level transformations;IMDB movie reviews;Enron spam emails;real-world text datasets;scoring strategies;text input;text perturbations;DeepWordBug;black-box attack;adversarial text sequences;black-box generation;Perturbation methods;Machine learning;Task analysis;Recurrent neural networks;Prediction algorithms;Sentiment analysis;adversarial samples;black box attack;text classification;misclassification;word embedding;deep learning},
doi={10.1109/SPW.2018.00016},
month={May},}
Having trouble with our tools? Please contact me and we’ll help you sort it out.
12 Dec 2017
About
We have designed and implemented EvadeML-Zoo, a benchmarking and visualization tool for research on adversarial machine learning. The goal of EvadeML-Zoo is to ease the experimental setup and help researchers evaluate and verify their results.
EvadeML-Zoo has a modular architecture and is designed to make it easy to add new datasets, pre-trained target models, attack or defense algorithms. The code is open source under the MIT license.
We have integrated three popular datasets: MNIST, CIFAR-10 and ImageNet- ILSVRC with a simple and unified interface. We offer several representative pre-trained models with state-of-the-art accuracy for each dataset including two pre-trained models for ImageNet-ILSVRC: the heavy Inception-v3 and and the lightweight MobileNet. We use Keras to access the pre-trained models because it provides a simplified interface and it is compatible with TensorFlow, which is a flexible tool for implementing attack and defense techniques.
We have integrated several existing attack algorithms as baseline for the upcoming new methods, including FGSM, BIM, JSMA, Deepfool, Universal Adversarial Perturbations, and Carlini and Wagner’s algorithms.
We have integrated our “feature squeezing” based detection framework in this toolbox. Formulating detecting adversarial examples as a binary classification task, we first construct a balanced dataset with equal number of legitimate and adversarial examples, and then split it into training and test subsets. A detection method has full access to the training set but no access to the labels of the test set. We measure the TPR and FPR on the test set as the benchmark detection results. Our Feature Squeezing functions as the detection baseline. Users can easily add more detection methods using our framework.
Besides, the tool comes with an interactive web-based visualization module adapted from our previous ADVERSARIAL-PLAYGROUND package. This module enables better understanding of the impact of attack algorithms on the resulting adversarial sample; users may specify attack algorithm parameters for a variety of attack types and generate new samples on-demand. The interface displays the resulting adversarial example as compared to the original, classification likelihoods, and the influence of a target model throughout layers of the network.
Citations
@inproceedings{Xu0Q18,
author = {Weilin Xu and
David Evans and
Yanjun Qi},
title = {Feature Squeezing: Detecting Adversarial Examples in Deep Neural Networks},
booktitle = {25th Annual Network and Distributed System Security Symposium, {NDSS}
2018, San Diego, California, USA, February 18-21, 2018},
year = {2018},
crossref = {DBLP:conf/ndss/2018},
url = {http://wp.internetsociety.org/ndss/wp-content/uploads/sites/25/2018/02/ndss2018\_03A-4\_Xu\_paper.pdf},
timestamp = {Thu, 09 Aug 2018 10:57:16 +0200},
biburl = {https://dblp.org/rec/bib/conf/ndss/Xu0Q18},
bibsource = {dblp computer science bibliography, https://dblp.org}
}
Having troubl with our tools? Please contact Weilin and we’ll help you sort it out.
11 Dec 2017
Jack’s DeepMotif paper (Deep Motif Dashboard: Visualizing and Understanding Genomic Sequences Using Deep Neural Networks ) have received the “best paper award“ at NIPS17 workshop for Transparent and interpretable Machine Learning in Safety Critical Environments. Big congratulations!!!
10 Dec 2017
Prototype Matching Networks : A novel deep learning architecture for Large-Scale Multi-label Genomic Sequence Classification
Abstract
One of the fundamental tasks in understanding genomics is the problem of predicting Transcription Factor Binding Sites (TFBSs). With more than hundreds of Transcription Factors (TFs) as labels, genomic-sequence based TFBS prediction is a challenging multi-label classification task. There are two major biological mechanisms for TF binding: (1) sequence-specific binding patterns on genomes known as “motifs” and (2) interactions among TFs known as co-binding effects. In this paper, we propose a novel deep architecture, the Prototype Matching Network (PMN) to mimic the TF binding mechanisms. Our PMN model automatically extracts prototypes (“motif”-like features) for each TF through a novel prototype-matching loss. Borrowing ideas from few-shot matching models, we use the notion of support set of prototypes and an LSTM to learn how TFs interact and bind to genomic sequences. On a reference TFBS dataset with 2.1 million genomic sequences, PMN significantly outperforms baselines and validates our design choices empirically. To our knowledge, this is the first deep learning architecture that introduces prototype learning and considers TF-TF interactions for large-scale TFBS prediction. Not only is the proposed architecture accurate, but it also models the underlying biology.
Citations
@article{lanchantin2017prototype,
title={Prototype Matching Networks for Large-Scale Multi-label Genomic Sequence Classification},
author={Lanchantin, Jack and Sekhon, Arshdeep and Singh, Ritambhara and Qi, Yanjun},
journal={arXiv preprint arXiv:1710.11238},
year={2017}
}
Having trouble with our tools? Please contact Jack and we’ll help you sort it out.
08 Nov 2017
Presentation: Slides @ AISTAT18
Poster @ NIPS 2017 workshop for Advances in Modeling and Learning Interactions from Complex Data.
R package: CRAN
install.packages("diffee")
library(diffee)
demo(diffee)
Abstract
We focus on the problem of estimating the change in the dependency structures of two p-dimensional Gaussian Graphical models (GGMs). Previous studies for sparse change estimation in GGMs involve expensive and difficult non-smooth optimization. We propose a novel method, DIFFEE for estimating DIFFerential networks via an Elementary Estimator under a high-dimensional situation. DIFFEE is solved through a faster and closed form solution that enables it to work in large-scale settings. We conduct a rigorous statistical analysis showing that surprisingly DIFFEE achieves the same asymptotic convergence rates as the state-of-the-art estimators that are much more difficult to compute. Our experimental results on multiple synthetic datasets and one real-world data about brain connectivity show strong performance improvements over baselines, as well as significant computational benefits.
Citations
@InProceedings{pmlr-v84-wang18f,
title = {Fast and Scalable Learning of Sparse Changes in High-Dimensional Gaussian Graphical Model Structure},
author = {Beilun Wang and arshdeep Sekhon and Yanjun Qi},
booktitle = {Proceedings of the Twenty-First International Conference on Artificial Intelligence and Statistics},
pages = {1691--1700},
year = {2018},
editor = {Amos Storkey and Fernando Perez-Cruz},
volume = {84},
series = {Proceedings of Machine Learning Research},
address = {Playa Blanca, Lanzarote, Canary Islands},
month = {09--11 Apr},
publisher = {PMLR},
pdf = {http://proceedings.mlr.press/v84/wang18f/wang18f.pdf},
url = {http://proceedings.mlr.press/v84/wang18f.html},
abstract = {We focus on the problem of estimating the change in the dependency structures of two $p$-dimensional Gaussian Graphical models (GGMs). Previous studies for sparse change estimation in GGMs involve expensive and difficult non-smooth optimization. We propose a novel method, DIFFEE for estimating DIFFerential networks via an Elementary Estimator under a high-dimensional situation. DIFFEE is solved through a faster and closed form solution that enables it to work in large-scale settings. We conduct a rigorous statistical analysis showing that surprisingly DIFFEE achieves the same asymptotic convergence rates as the state-of-the-art estimators that are much more difficult to compute. Our experimental results on multiple synthetic datasets and one real-world data about brain connectivity show strong performance improvements over baselines, as well as significant computational benefits.}
}
Having trouble with our tools? Please contact Beilun and we’ll help you sort it out.
08 Sep 2017
We are updating the R package: simule with one more function: W-SIMULE
install.packages("simule")
library(simule)
demo(wsimule)
Paper: @Arxiv @ NIPS 2017 workshop for Advances in Modeling and Learning Interactions from Complex Data.
Abstract
Determining functional brain connectivity is crucial to understanding the brain and neural differences underlying disorders such as autism. Recent studies have used Gaussian graphical models to learn brain connectivity via statistical dependencies across brain regions from neuroimaging. However, previous studies often fail to properly incorporate priors tailored to neuroscience, such as preferring shorter connections. To remedy this problem, the paper here introduces a novel, weighted-ℓ1, multi-task graphical model (W-SIMULE). This model elegantly incorporates a flexible prior, along with a parallelizable formulation. Additionally, W-SIMULE extends the often-used Gaussian assumption, leading to considerable performance increases. Here, applications to fMRI data show that W-SIMULE succeeds in determining functional connectivity in terms of (1) log-likelihood, (2) finding edges that differentiate groups, and (3) classifying different groups based on their connectivity, achieving 58.6\% accuracy on the ABIDE dataset. Having established W-SIMULE’s effectiveness, it links four key areas to autism, all of which are consistent with the literature. Due to its elegant domain adaptivity, W-SIMULE can be readily applied to various data types to effectively estimate connectivity.

Citations
@article{singh2017constrained,
title={A Constrained, Weighted-L1 Minimization Approach for Joint Discovery of Heterogeneous Neural Connectivity Graphs},
author={Singh, Chandan and Wang, Beilun and Qi, Yanjun},
journal={arXiv preprint arXiv:1709.04090},
year={2017}
}
Having trouble with our tools? Please contact Beilun and we’ll help you sort it out.
03 Aug 2017
Revised Version2 Paper Arxiv
Revised Title: Adversarial-Playground: A Visualization Suite Showing How Adversarial Examples Fool Deep Learning
Publish @ The IEEE Symposium on Visualization for Cyber Security (VizSec) 2017 -ULR

Abstract
Recent studies have shown that attackers can force deep learning models to
misclassify so-called “adversarial examples”: maliciously generated images
formed by making imperceptible modifications to pixel values. With growing
interest in deep learning for security applications, it is important for
security experts and users of machine learning to recognize how learning
systems may be attacked. Due to the complex nature of deep learning, it is
challenging to understand how deep models can be fooled by adversarial
examples. Thus, we present a web-based visualization tool,
Adversarial-Playground, to demonstrate the efficacy of common adversarial
methods against a convolutional neural network (CNN) system.
Adversarial-Playground is educational, modular and interactive. (1) It enables
non-experts to compare examples visually and to understand why an adversarial
example can fool a CNN-based image classifier. (2) It can help security experts
explore more vulnerability of deep learning as a software module. (3) Building
an interactive visualization is challenging in this domain due to the large
feature space of image classification (generating adversarial examples is slow
in general and visualizing images are costly). Through multiple novel design
choices, our tool can provide fast and accurate responses to user requests.
Empirically, we find that our client-server division strategy reduced the
response time by an average of 1.5 seconds per sample. Our other innovation, a
faster variant of JSMA evasion algorithm, empirically performed twice as fast
as JSMA and yet maintains a comparable evasion rate.
Project source code and data from our experiments available at:
GitHub
Citations
@inproceedings{norton2017adversarial,
title={Adversarial-Playground: A visualization suite showing how adversarial examples fool deep learning},
author={Norton, Andrew P and Qi, Yanjun},
booktitle={Visualization for Cyber Security (VizSec), 2017 IEEE Symposium on},
pages={1--4},
year={2017},
organization={IEEE}
}
Having trouble with our tools? Please contact Andrew Norton and we’ll help you sort it out.
02 Aug 2017
Abstract
Feature squeezing is a recently-introduced framework for mitigating and detecting adversarial examples. In previous work, we showed that it is effective against several earlier methods for generating adversarial examples. In this short note, we report on recent results showing that simple feature squeezing techniques also make deep learning models significantly more robust against the Carlini/Wagner attacks, which are the best known adversarial methods discovered to date.
Citations
@article{xu2017feature,
title={Feature Squeezing Mitigates and Detects Carlini/Wagner Adversarial Examples},
author={Xu, Weilin and Evans, David and Qi, Yanjun},
journal={arXiv preprint arXiv:1705.10686},
year={2017}
}
Having troubl with our tools? Please contact Weilin and we’ll help you sort it out.
30 Jul 2017
Paper: @Arxiv + Published at [NIPS2017]
(https://papers.nips.cc/paper/7255-attend-and-predict-understanding-gene-regulation-by-selective-attention-on-chromatin.pdf)
Abstract:
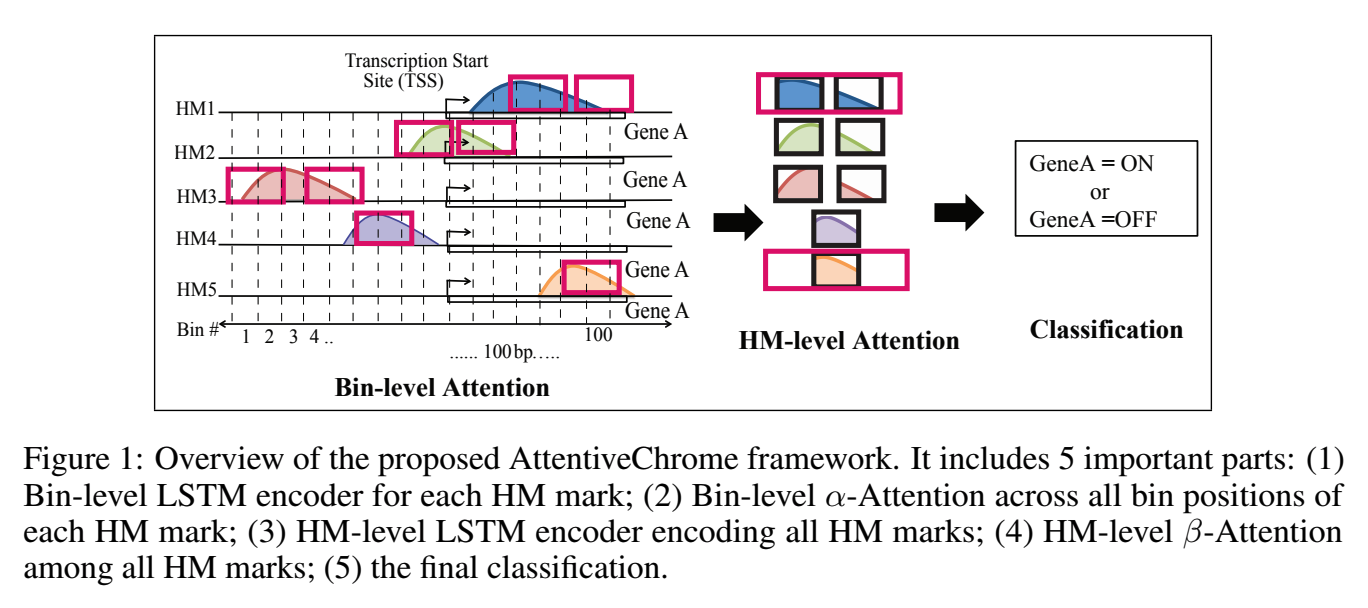
The past decade has seen a revolution in genomic technologies that enable a flood of genome-wide profiling of chromatin marks. Recent literature tried to understand gene regulation by predicting gene expression from large-scale chromatin measurements. Two fundamental challenges exist for such learning tasks: (1) genome-wide chromatin signals are spatially structured, high-dimensional and highly modular; and (2) the core aim is to understand what are the relevant factors and how they work together? Previous studies either failed to model complex dependencies among input signals or relied on separate feature analysis to explain the decisions. This paper presents an attention-based deep learning approach; we call AttentiveChrome, that uses a unified architecture to model and to interpret dependencies among chromatin factors for controlling gene regulation. AttentiveChrome uses a hierarchy of multiple Long short-term memory (LSTM) modules to encode the input signals and to model how various chromatin marks cooperate automatically. AttentiveChrome trains two levels of attention jointly with the target prediction, enabling it to attend differentially to relevant marks and to locate important positions per mark. We evaluate the model across 56 different cell types (tasks) in human. Not only is the proposed architecture more accurate, but its attention scores also provide a better interpretation than state-of-the-art feature visualization methods such as saliency map.
Code and data are shared at www.deepchrome.net
Citations
@inproceedings{singh2017attend,
title={Attend and Predict: Understanding Gene Regulation by Selective Attention on Chromatin},
author={Singh, Ritambhara and Lanchantin, Jack and Sekhon, Arshdeep and Qi, Yanjun},
booktitle={Advances in Neural Information Processing Systems},
pages={6769--6779},
year={2017}
}
Having trouble with our tools? Please contact Rita and we’ll help you sort it out.
12 Jun 2017
Abstract
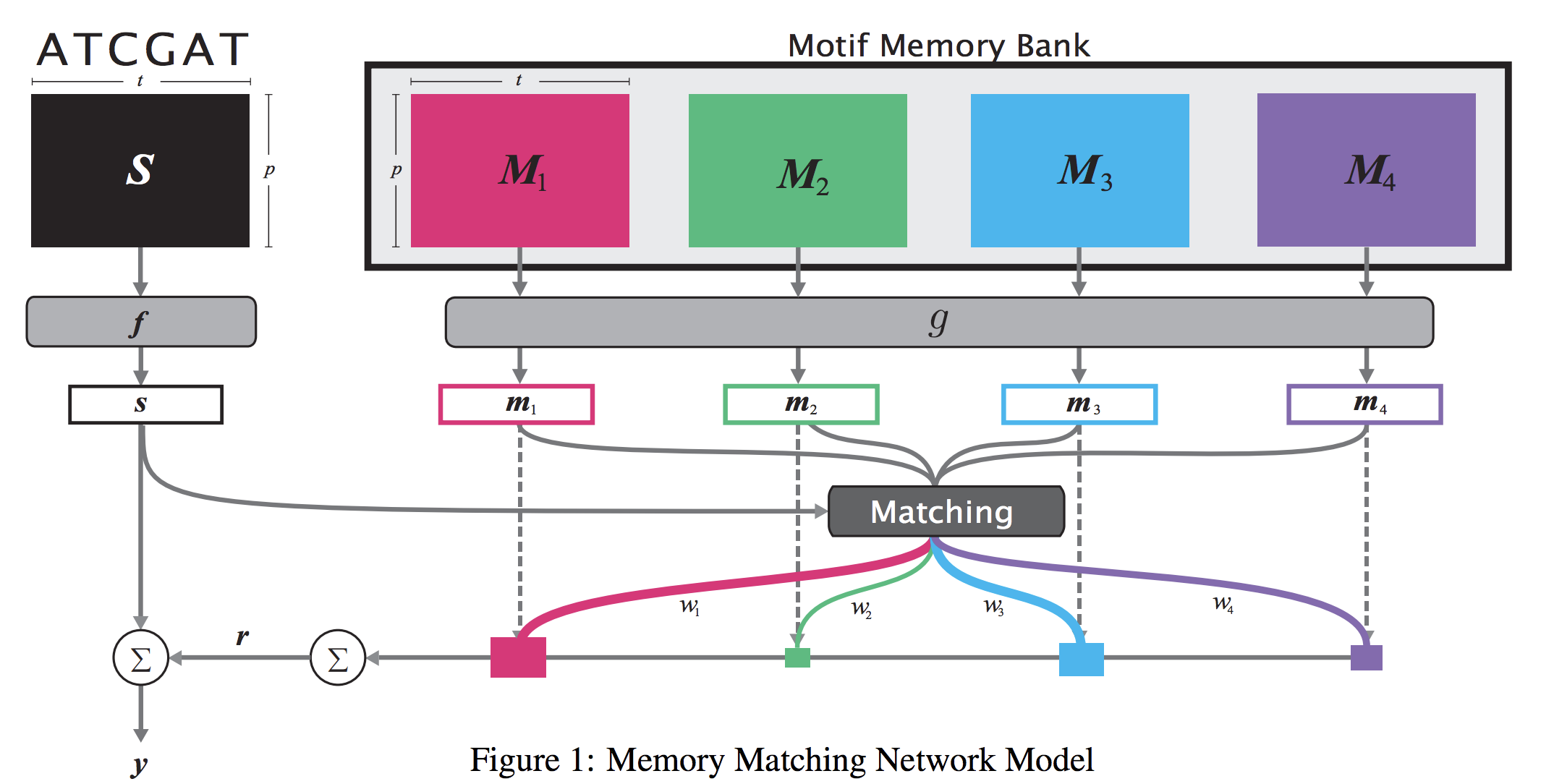
When analyzing the genome, researchers have discovered that proteins bind to DNA based on certain patterns of the DNA sequence known as “motifs”. However, it is difficult to manually construct motifs due to their complexity. Recently, externally learned memory models have proven to be effective methods for reasoning over inputs and supporting sets. In this work, we present memory matching networks (MMN) for classifying DNA sequences as protein binding sites. Our model learns a memory bank of encoded motifs, which are dynamic memory modules, and then matches a new test sequence to each of the motifs to classify the sequence as a binding or nonbinding site.
Citations
@article{lanchantin2017memory,
title={Memory Matching Networks for Genomic Sequence Classification},
author={Lanchantin, Jack and Singh, Ritambhara and Qi, Yanjun},
journal={arXiv preprint arXiv:1702.06760},
year={2017}
}
Having trouble with our tools? Please contact Jack and we’ll help you sort it out.
11 Jun 2017
Abstract:
Deep neural network (DNN) models have recently obtained state-of-the-art prediction accuracy for the transcription factor binding (TFBS) site classification task. However, it remains unclear how these approaches identify meaningful DNA sequence signals and give insights as to why TFs bind to certain locations. In this paper, we propose a toolkit called the Deep Motif Dashboard (DeMo Dashboard) which provides a suite of visualization strategies to extract motifs, or sequence patterns from deep neural network models for TFBS classification. We demonstrate how to visualize and understand three important DNN models: convolutional, recurrent, and convolutional-recurrent networks. Our first visualization method is finding a test sequence’s saliency map which uses first-order derivatives to describe the importance of each nucleotide in making the final prediction. Second, considering recurrent models make predictions in a temporal manner (from one end of a TFBS sequence to the other), we introduce temporal output scores, indicating the prediction score of a model over time for a sequential input. Lastly, a class-specific visualization strategy finds the optimal input sequence for a given TFBS positive class via stochastic gradient optimization. Our experimental results indicate that a convolutional-recurrent architecture performs the best among the three architectures. The visualization techniques indicate that CNN-RNN makes predictions by modeling both motifs as well as dependencies among them.
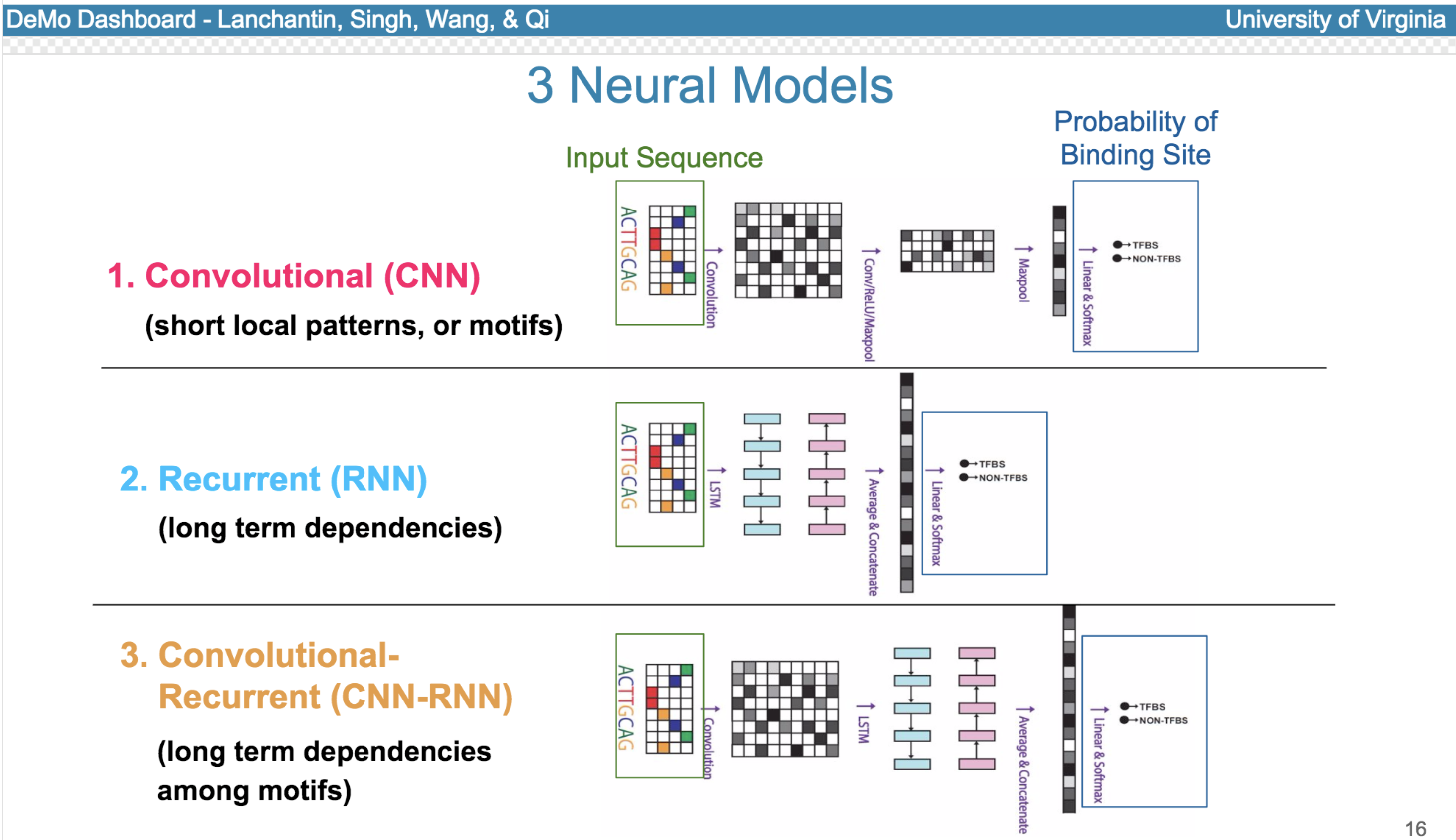
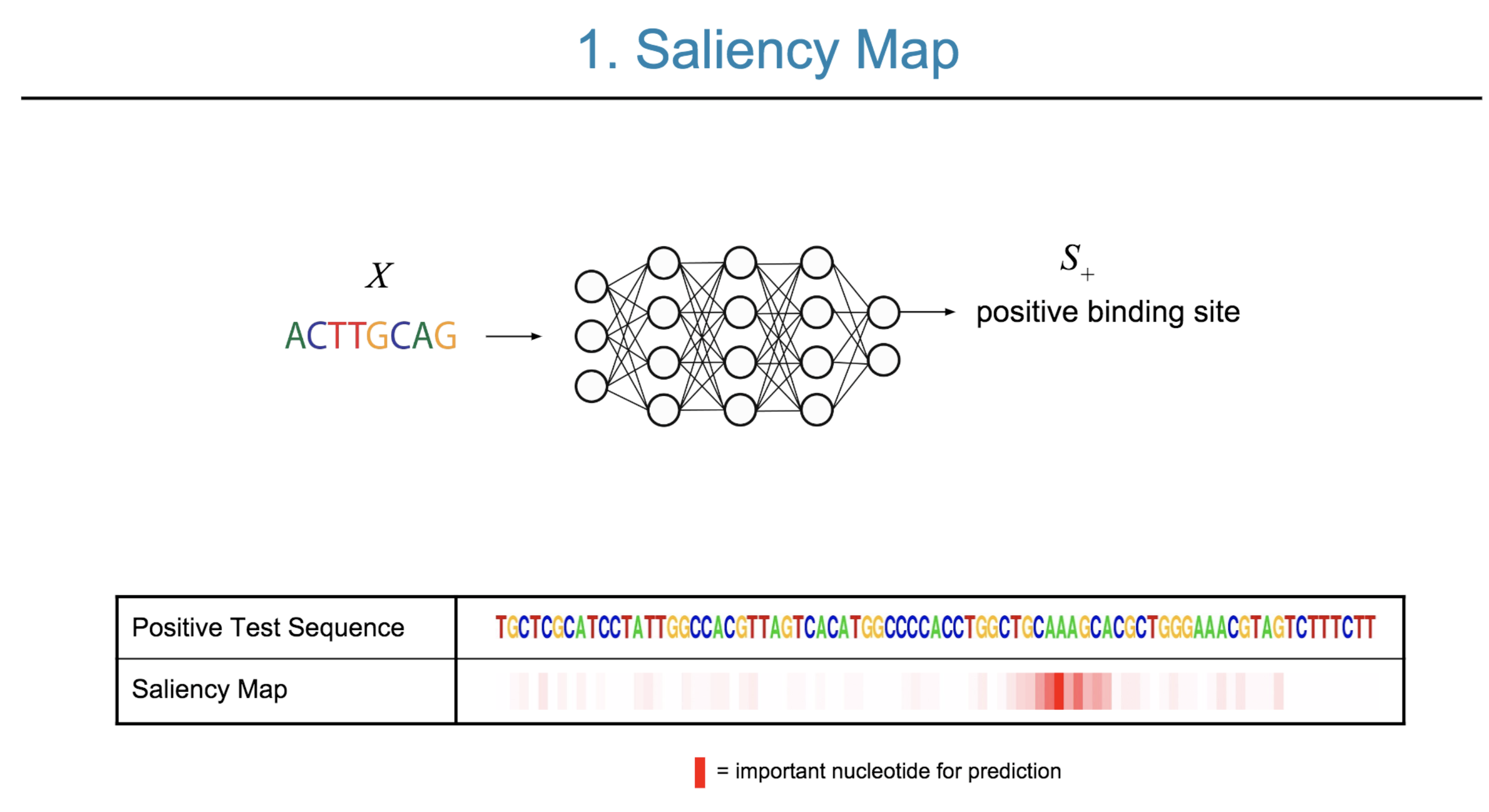
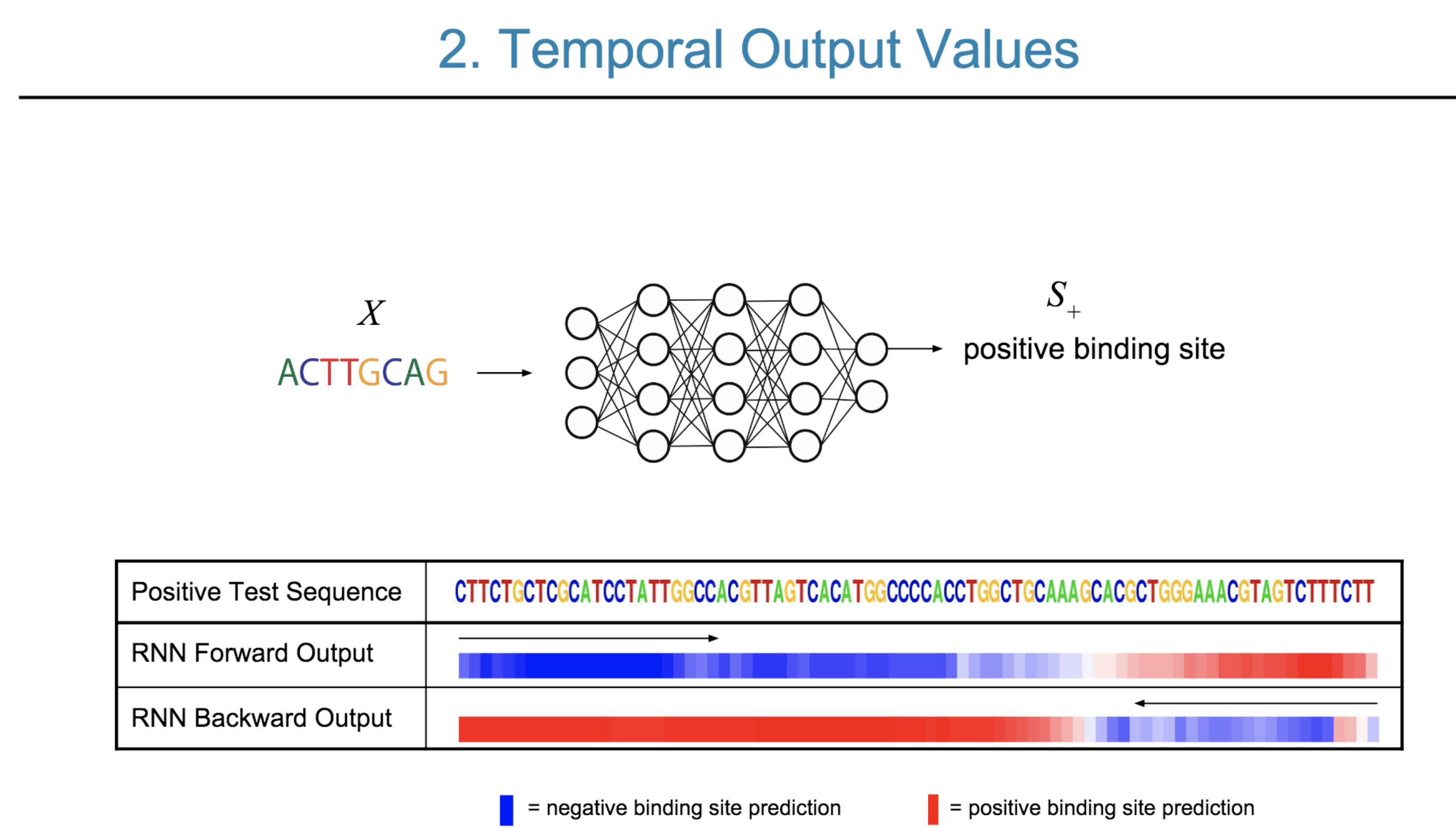
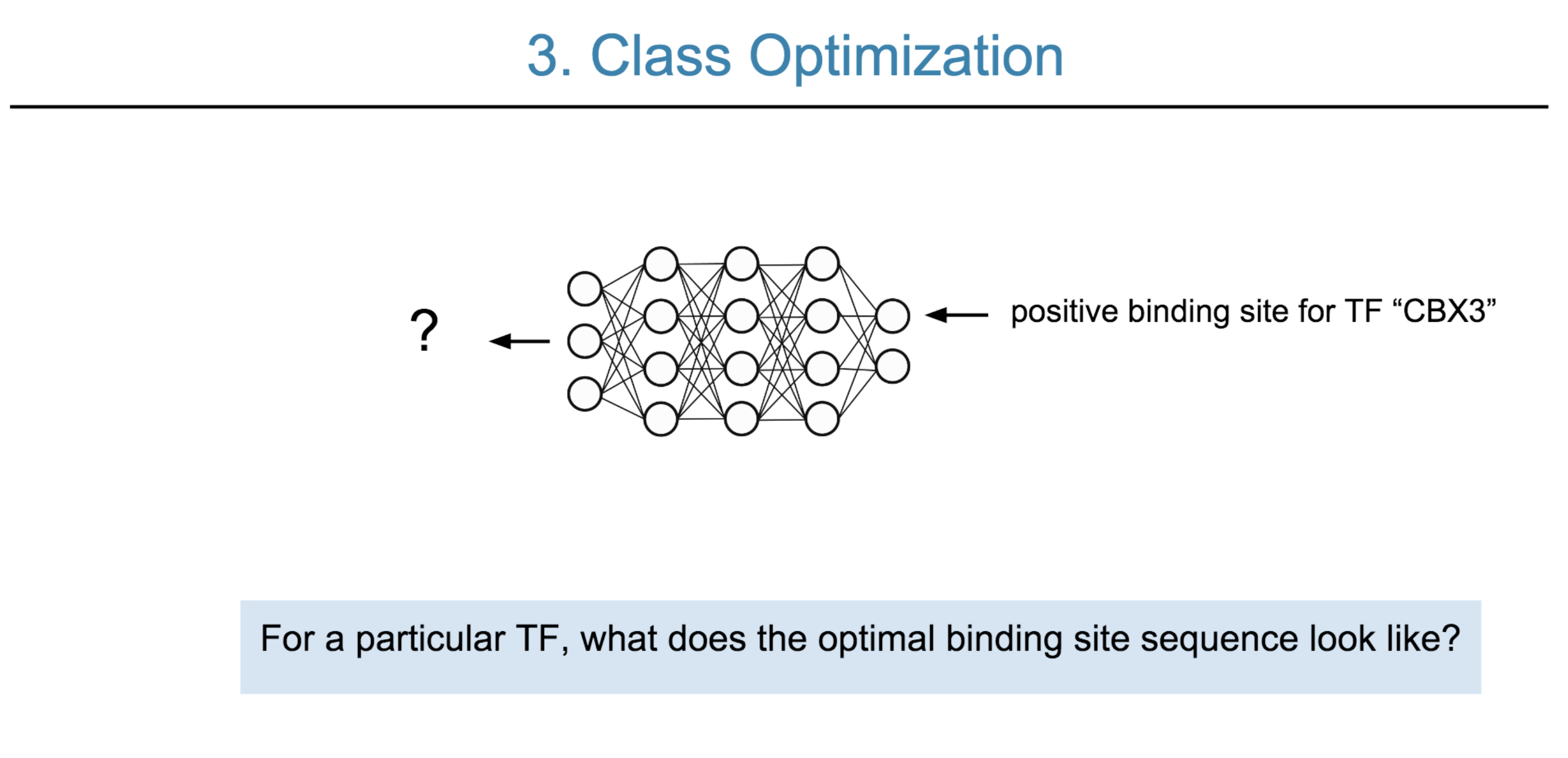
Citations
@inproceedings{lanchantin2017deep,
title={Deep motif dashboard: Visualizing and understanding genomic sequences using deep neural networks},
author={Lanchantin, Jack and Singh, Ritambhara and Wang, Beilun and Qi, Yanjun},
booktitle={PACIFIC SYMPOSIUM ON BIOCOMPUTING 2017},
pages={254--265},
year={2017},
organization={World Scientific}
}
Having trouble with our tools? Please contact Jack and we’ll help you sort it out.
10 Jun 2017
Abstract:
Motivation: Histone modifications are among the most important factors that control gene regulation. Computational methods that predict gene expression from histone modification signals are highly desirable for understanding their combinatorial effects in gene regulation. This knowledge can help in developing ‘epigenetic drugs’ for diseases like cancer. Previous studies for quantifying the relationship between histone modifications and gene expression levels either failed to capture combinatorial effects or relied on multiple methods that separate predictions and combinatorial analysis. This paper develops a unified discriminative framework using a deep convolutional neural network to classify gene expression using histone modification data as input. Our system, called DeepChrome, allows automatic extraction of complex interactions among important features. To simultaneously visualize the combinatorial interactions among histone modifications, we propose a novel optimization-based technique that generates feature pattern maps from the learnt deep model. This provides an intuitive description of underlying epigenetic mechanisms that regulate genes. Results: We show that DeepChrome outperforms state-of-the-art models like Support Vector Machines and Random Forests for gene expression classification task on 56 different cell-types from REMC database. The output of our visualization technique not only validates the previous observations but also allows novel insights about combinatorial interactions among histone modification marks, some of which have recently been observed by experimental studies.
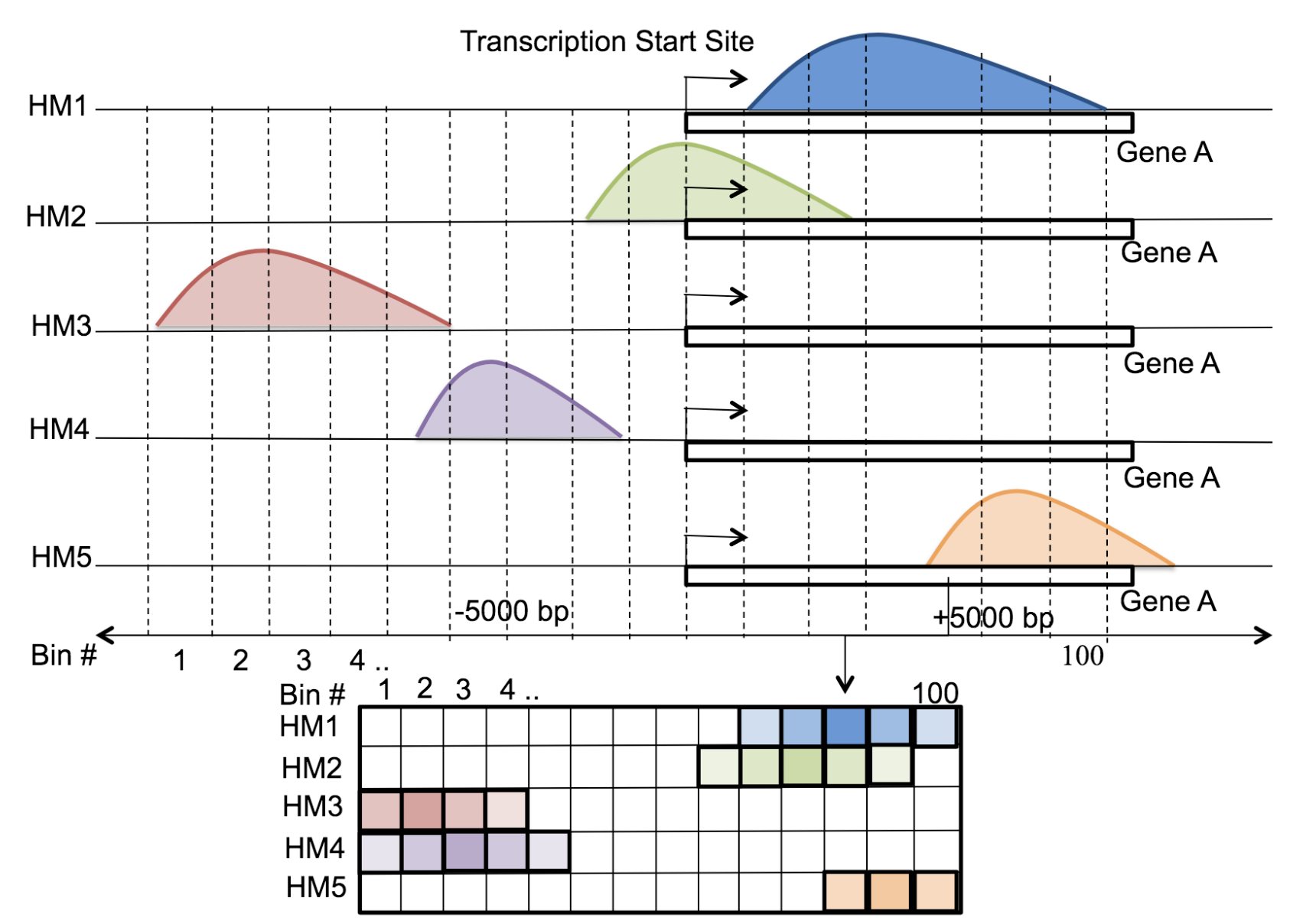
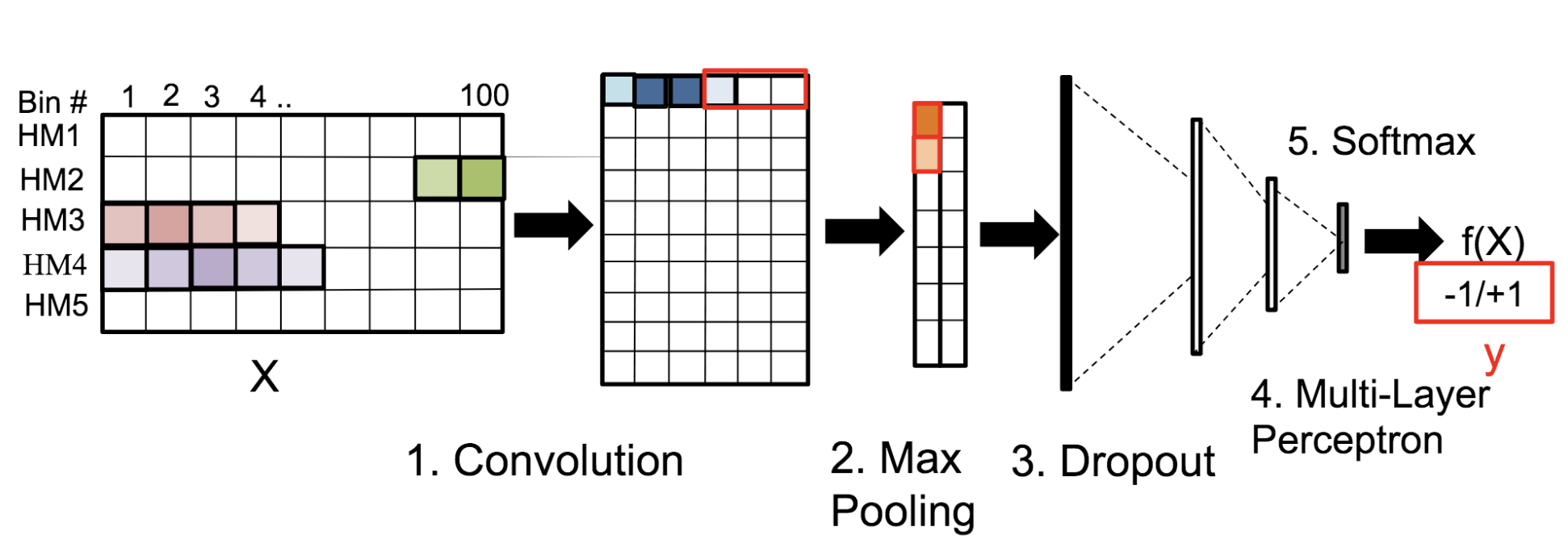
Citations
@article{singh2016deepchrome,
title={DeepChrome: deep-learning for predicting gene expression from histone modifications},
author={Singh, Ritambhara and Lanchantin, Jack and Robins, Gabriel and Qi, Yanjun},
journal={Bioinformatics},
volume={32},
number={17},
pages={i639--i648},
year={2016},
publisher={Oxford University Press}
}
Having trouble with our tools? Please contact Rita and we’ll help you sort it out.
04 Jun 2017
Abstract
With growing interest in adversarial machine learning, it is important for machine learning practitioners and users to understand how their models may be attacked. We propose a web-based visualization tool, \textit{Adversarial-Playground}, to demonstrate the efficacy of common adversarial methods against a deep neural network (DNN) model, built on top of the TensorFlow library. Adversarial-Playground provides users an efficient and effective experience in exploring techniques generating adversarial examples, which are inputs crafted by an adversary to fool a machine learning system. To enable Adversarial-Playground to generate quick and accurate responses for users, we use two primary tactics: (1) We propose a faster variant of the state-of-the-art Jacobian saliency map approach that maintains a comparable evasion rate. (2) Our visualization does not transmit the generated adversarial images to the client, but rather only the matrix describing the sample and the vector representing classification likelihoods.
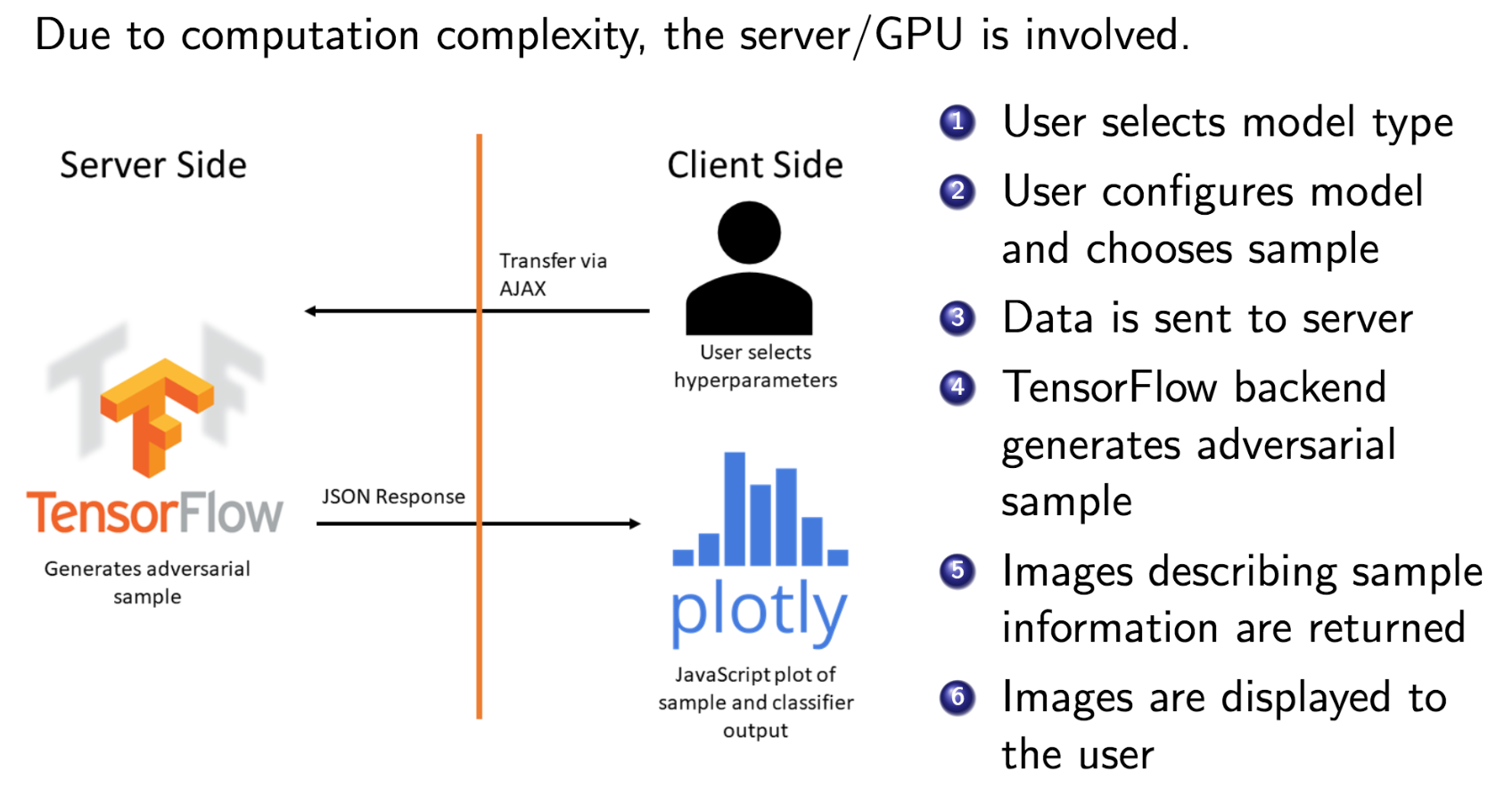

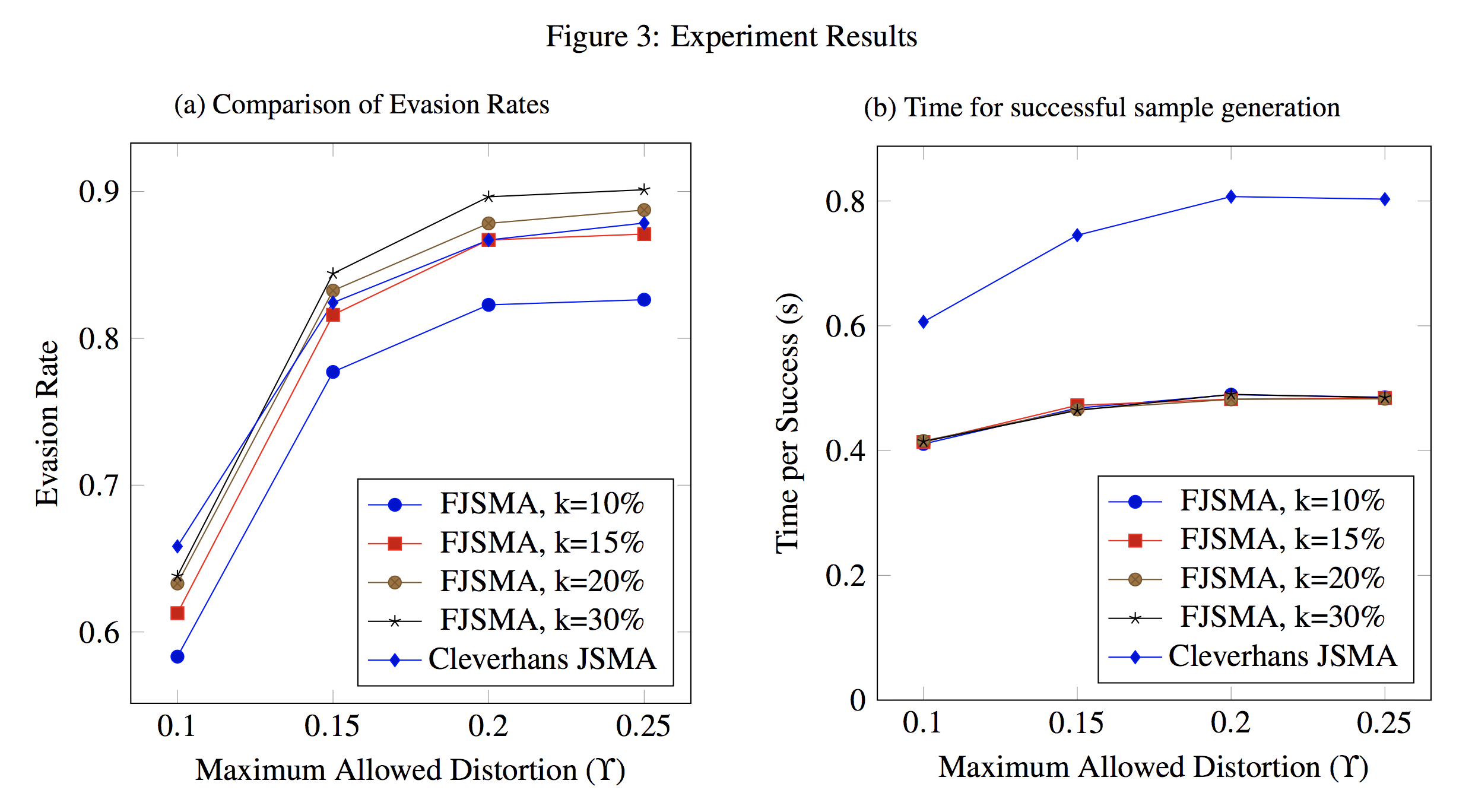
Citations
@inproceedings{norton2017adversarial,
title={Adversarial-Playground: A visualization suite showing how adversarial examples fool deep learning},
author={Norton, Andrew P and Qi, Yanjun},
booktitle={Visualization for Cyber Security (VizSec), 2017 IEEE Symposium on},
pages={1--4},
year={2017},
organization={IEEE}
}
Having trouble with our tools? Please contact Andrew Norton and we’ll help you sort it out.
03 Jun 2017
Abstract
Recent studies have shown that deep neural networks (DNN) are vulnerable to adversarial samples: maliciously-perturbed samples crafted to yield incorrect model outputs. Such attacks can severely undermine DNN systems, particularly in security-sensitive settings. It was observed that an adversary could easily generate adversarial samples by making a small perturbation on irrelevant feature dimensions that are unnecessary for the current classification task. To overcome this problem, we introduce a defensive mechanism called DeepCloak. By identifying and removing unnecessary features in a DNN model, DeepCloak limits the capacity an attacker can use generating adversarial samples and therefore increase the robustness against such inputs. Comparing with other defensive approaches, DeepCloak is easy to implement and computationally efficient. Experimental results show that DeepCloak can increase the performance of state-of-the-art DNN models against adversarial samples.
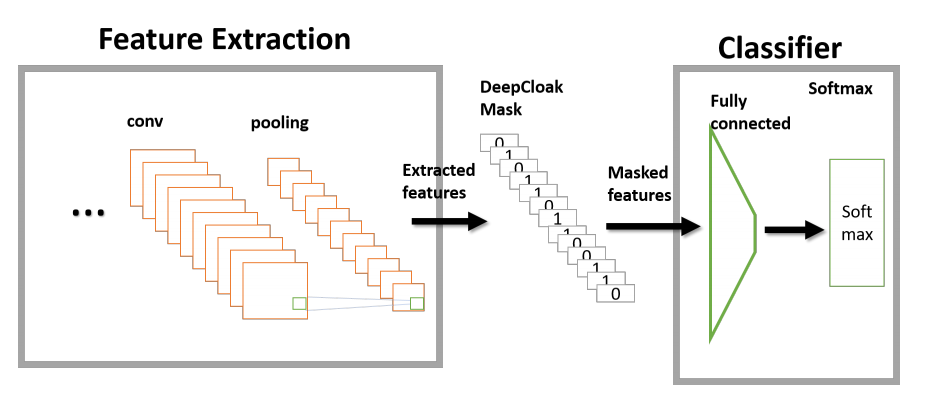
Citations
@article{gao2017deepmask,
title={DeepCloak: Masking DNN Models for robustness against adversarial samples},
author={Gao, Ji and Wang, Beilun and Qi, Yanjun},
journal={arXiv preprint arXiv:1702.06763},
year={2017}
}
Having trouble with our tools? Please contact Ji Gao and we’ll help you sort it out.
01 Jun 2017
Paper: Automatically Evading Classifiers,
A Case Study on PDF Malware Classifiers NDSS16
More information is provided by EvadeML.org
By using evolutionary techniques to simulate an adversary’s efforts to evade that classifier
Abstract
Machine learning is widely used to develop classifiers for security tasks. However, the robustness of these methods
against motivated adversaries is uncertain. In this work, we
propose a generic method to evaluate the robustness of classifiers
under attack. The key idea is to stochastically manipulate a
malicious sample to find a variant that preserves the malicious
behavior but is classified as benign by the classifier. We present
a general approach to search for evasive variants and report on
results from experiments using our techniques against two PDF
malware classifiers, PDFrate and Hidost. Our method is able to
automatically find evasive variants for both classifiers for all of
the 500 malicious seeds in our study. Our results suggest a general
method for evaluating classifiers used in security applications, and
raise serious doubts about the effectiveness of classifiers based
on superficial features in the presence of adversaries.
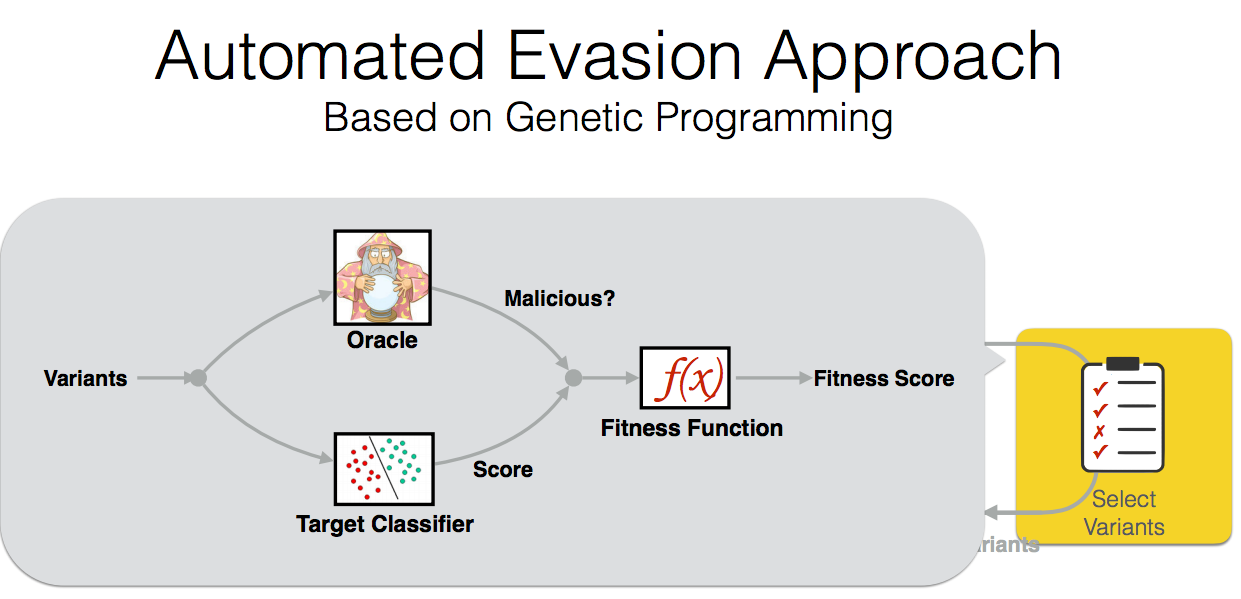
Citations
@inproceedings{xu2016automatically,
title={Automatically evading classifiers},
author={Xu, Weilin and Qi, Yanjun and Evans, David},
booktitle={Proceedings of the 2016 Network and Distributed Systems Symposium},
year={2016}
}
Having troubl with our tools? Please contact Weilin and we’ll help you sort it out.
11 May 2017
Abstract
Most machine learning classifiers, including deep neural networks, are vulnerable to adversarial examples. Such inputs are typically generated by adding small but purposeful modifications that lead to incorrect outputs while imperceptible to human eyes. The goal of this paper is not to introduce a single method, but to make theoretical steps towards fully understanding adversarial examples. By using concepts from topology, our theoretical analysis brings forth the key reasons why an adversarial example can fool a classifier (f1) and adds its oracle (f2, like human eyes) in such analysis. By investigating the topological relationship between two (pseudo)metric spaces corresponding to predictor f1 and oracle f2, we develop necessary and sufficient conditions that can determine if f1 is always robust (strong-robust) against adversarial examples according to f2. Interestingly our theorems indicate that just one unnecessary feature can make f1 not strong-robust, and the right feature representation learning is the key to getting a classifier that is both accurate and strong-robust.
Recent studies are mostly empirical and provide little understanding
of why an adversary can fool machine learning models with adversarial examples. Several
important questions have not been answered yet:
- What makes a classifier always robust to adversarial examples?
- Which parts of a classifier influence its robustness against adversarial examples more, compared
with the rest?
- What is the relationship between a classifier’s generalization accuracy and its robustness against
adversarial examples?
- Why (many) DNN classifiers are not robust against adversarial examples ? How to improve?
This paper uses the following framework
-
to understand adversarial examples (by considering the role of oracle):
-
The following figure provides a simple case illustration explaining unnecessary features make a classifier vulnerable to adversarial examples:
-
The following figure tries to explain why DNN models are vulnerable to adversarial examples:
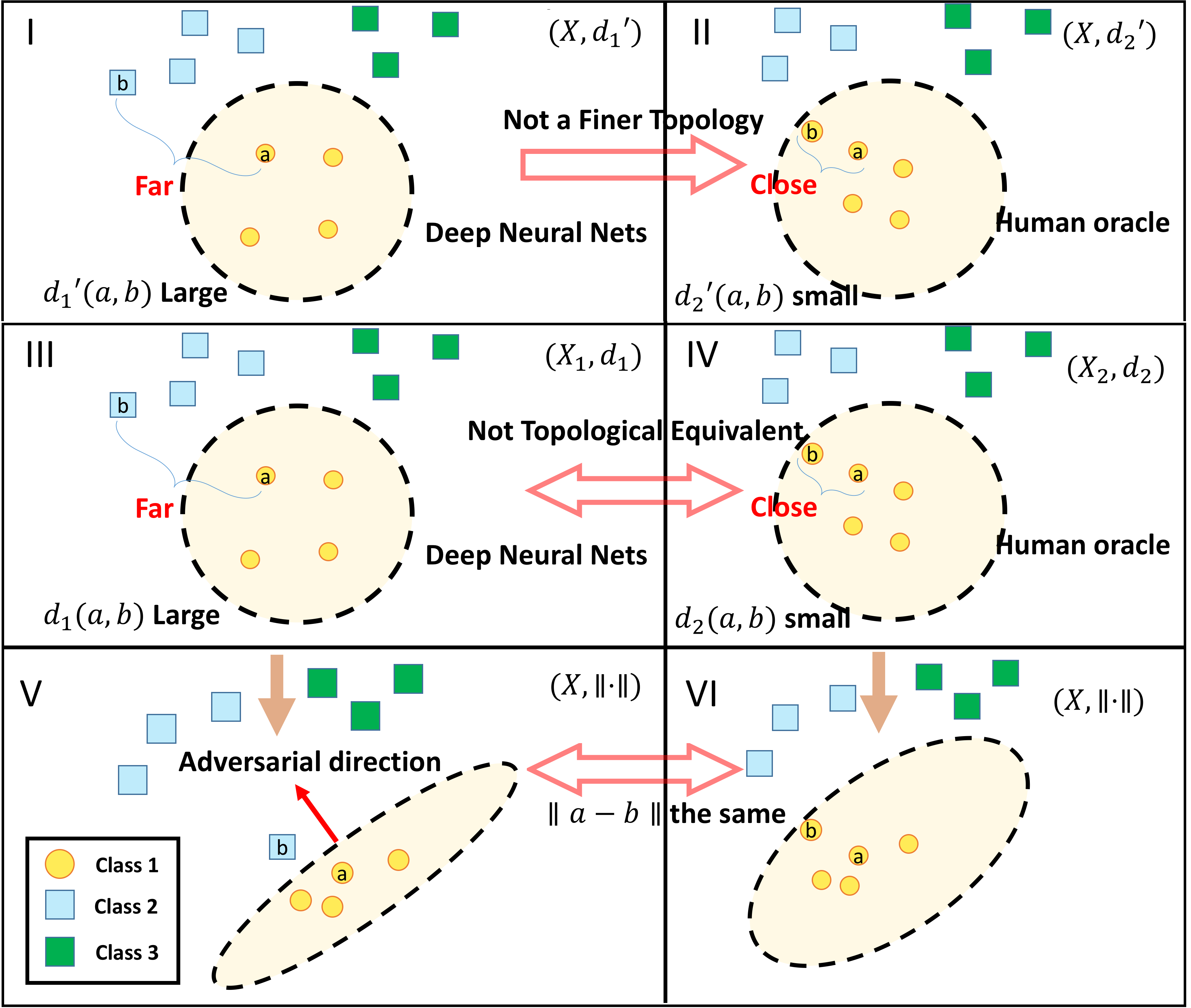
Citations
@article{wang2016theoretical,
title={A theoretical framework for robustness of (deep) classifiers under adversarial noise},
author={Wang, Beilun and Gao, Ji and Qi, Yanjun},
journal={arXiv preprint},
year={2016}
}
Having trouble with our tools? Please contact Beilun and we’ll help you sort it out.
20 Jun 2016
Abstract:
String Kernel (SK) techniques, especially those using gapped k-mers as features (gk), have obtained great success in classifying sequences like DNA, protein, and text. However, the state-of-the-art gk-SK runs extremely slow when we increase the dictionary size (Σ) or allow more mismatches (M). This is because current gk-SK uses a trie-based algorithm to calculate co-occurrence of mismatched substrings resulting in a time cost proportional to O(ΣM). We propose a \textbf{fast} algorithm for calculating \underline{Ga}pped k-mer \underline{K}ernel using \underline{Co}unting (GaKCo). GaKCo uses associative arrays to calculate the co-occurrence of substrings using cumulative counting. This algorithm is fast, scalable to larger Σ and M, and naturally parallelizable. We provide a rigorous asymptotic analysis that compares GaKCo with the state-of-the-art gk-SK. Theoretically, the time cost of GaKCo is independent of the ΣM term that slows down the trie-based approach. Experimentally, we observe that GaKCo achieves the same accuracy as the state-of-the-art and outperforms its speed by factors of 2, 100, and 4, on classifying sequences of DNA (5 datasets), protein (12 datasets), and character-based English text (2 datasets), respectively.
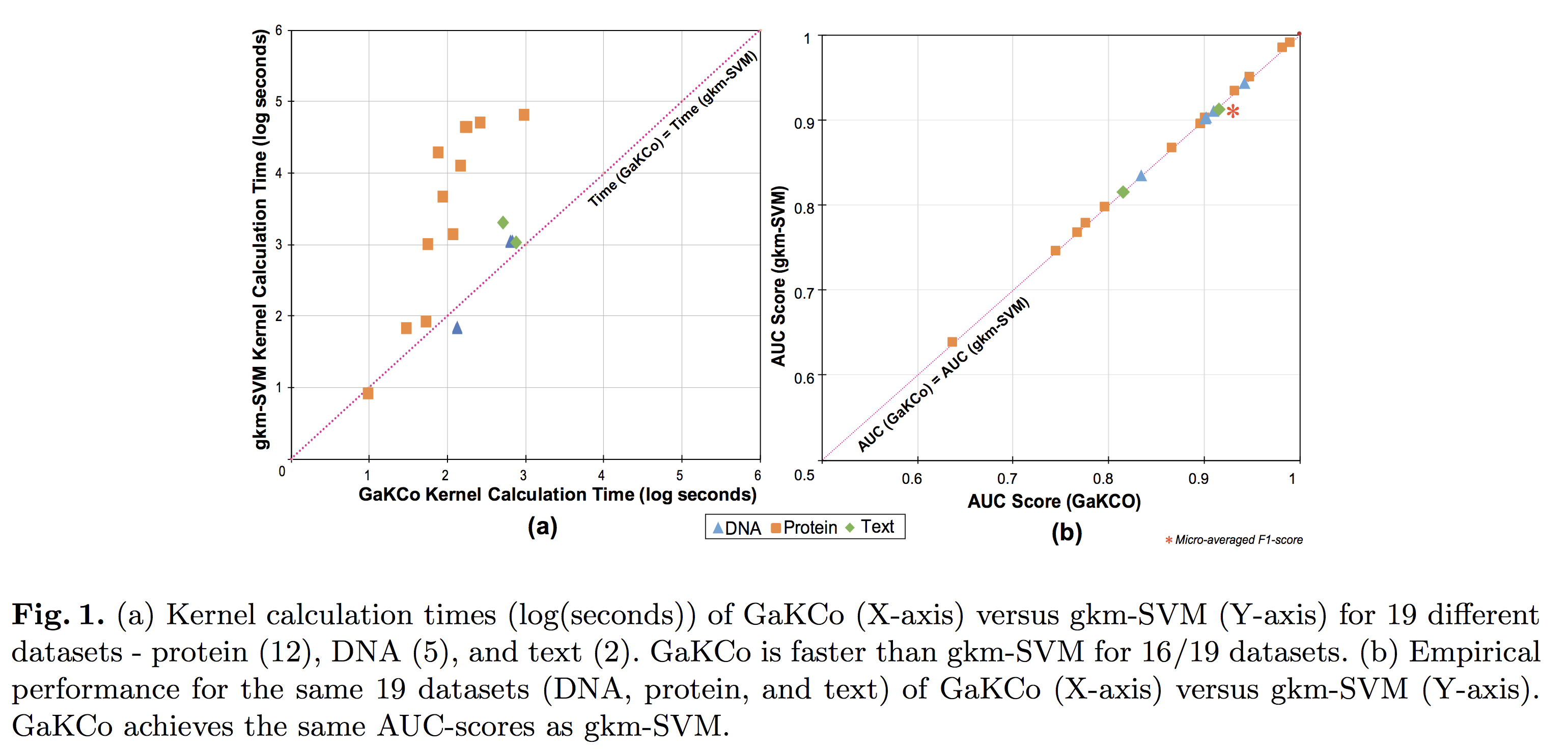
Citations
@inproceedings{singh_gakco:_2017,
location = {Cham},
title = {GaKCo: A Fast Gapped k-mer String Kernel Using Counting},
isbn = {978-3-319-71249-9},
pages = {356--373},
booktitle = {Machine Learning and Knowledge Discovery in Databases},
publisher = {Springer International Publishing},
author = {Singh, Ritambhara and Sekhon, Arshdeep and Kowsari, Kamran and Lanchantin, Jack and Wang, Beilun and Qi, Yanjun},
editor = {Ceci, Michelangelo and Hollmén, Jaakko and Todorovski, Ljupčo and Vens, Celine and Džeroski, Sašo},
date = {2017}
}
Having trouble with our tools? Please contact Rita and we’ll help you sort it out.
15 Jun 2016
Tool TSK: Transfer String Kernel for Cross-Context DNA-Protein Binding Prediction
Abstract
Through sequence-based classification, this paper tries to accurately predict the DNA binding sites of transcription factors (TFs) in an unannotated cellular context. Related methods in the literature fail to perform such predictions accurately, since they do not consider sample distribution shift of sequence segments from an annotated (source) context to an unannotated (target) context. We, therefore, propose a method called “Transfer String Kernel” (TSK) that achieves improved prediction of transcription factor binding site (TFBS) using knowledge transfer via cross-context sample adaptation. TSK maps sequence segments to a high-dimensional feature space using a discriminative mismatch string kernel framework. In this high-dimensional space, labeled examples of the source context are re-weighted so that the revised sample distribution matches the target context more closely. We have experimentally verified TSK for TFBS identifications on fourteen different TFs under a cross-organism setting. We find that TSK consistently outperforms the state-of the-art TFBS tools, especially when working with TFs whose binding sequences are not conserved across contexts. We also demonstrate the generalizability of TSK by showing its cutting-edge performance on a different set of cross-context tasks for the MHC peptide binding predictions.
Citations
@article{singh2016transfer,
title={Transfer String Kernel for Cross-Context DNA-Protein Binding Prediction},
author={Singh, Ritambhara and Lanchantin, Jack and Robins, Gabriel and Qi, Yanjun},
journal={IEEE/ACM Transactions on Computational Biology and Bioinformatics},
year={2016},
publisher={IEEE}
}
Having trouble with our tools? Please contact Rita and we’ll help you sort it out.
01 Jun 2016
install.packages("fasjem")
library(fasjem)
demo(fasjem)
Abstract
Estimating multiple sparse Gaussian Graphical
Models (sGGMs) jointly for many related
tasks (large K) under a high-dimensional
(large p) situation is an important task.
Most previous studies for the joint estimation
of multiple sGGMs rely on penalized
log-likelihood estimators that involve expensive
and difficult non-smooth optimizations.
We propose a novel approach, FASJEM for
fast and scalable joint structure-estimation of
multiple sGGMs at a large scale. As the first
study of joint sGGM using the M-estimator
framework, our work has three major contributions:
(1) We solve FASJEM through an
entry-wise manner which is parallelizable. (2)
We choose a proximal algorithm to optimize
FASJEM. This improves the computational
efficiency from O(Kp3
) to O(Kp2
) and reduces
the memory requirement from O(Kp2
)
to O(K). (3) We theoretically prove that FASJEM
achieves a consistent estimation with
a convergence rate of O(log(Kp)/ntot). On
several synthetic and four real-world datasets,
FASJEM shows significant improvements over
baselines on accuracy, computational complexity
and memory costs.

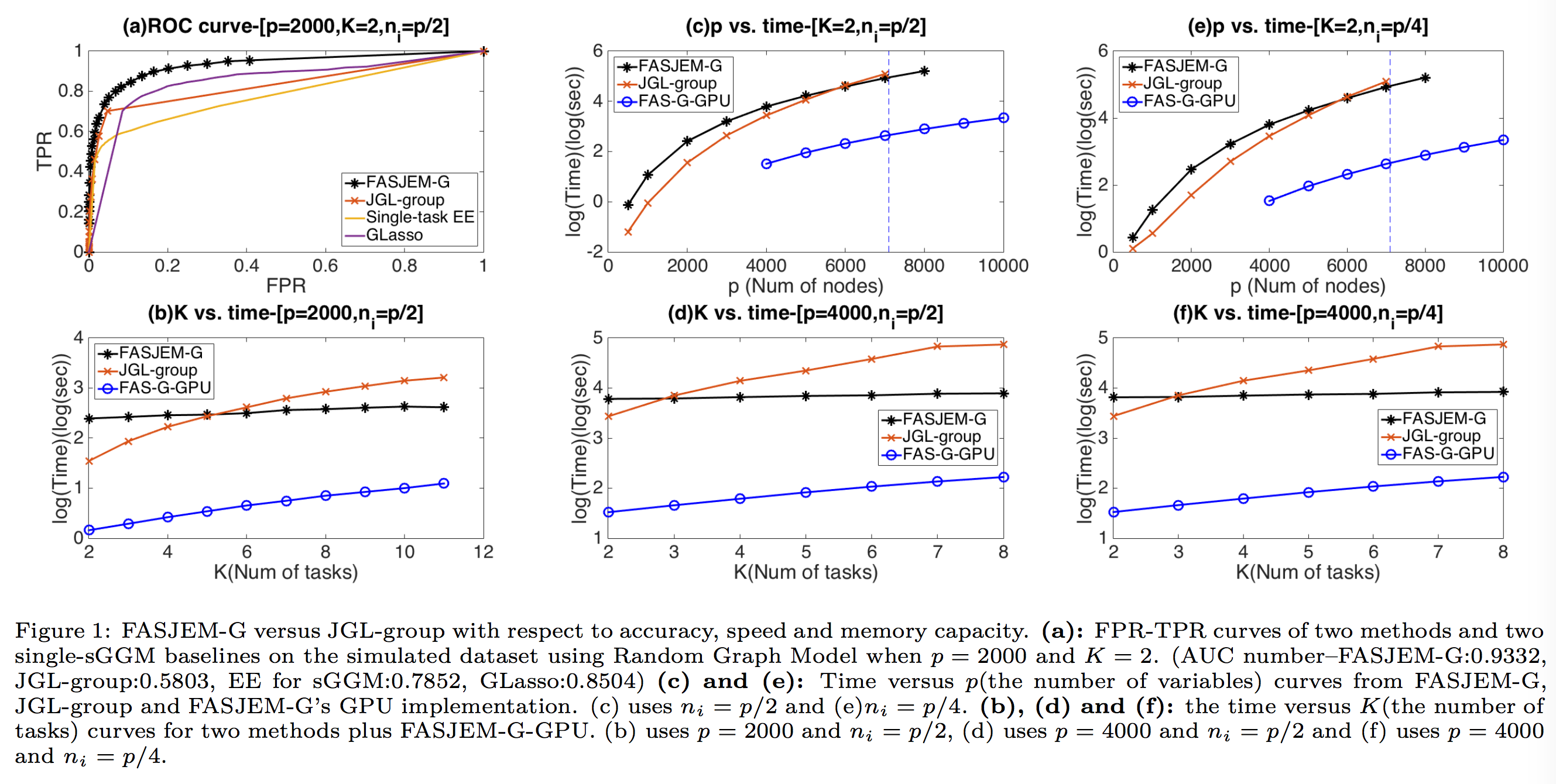
Citations
@inproceedings{wang2017fast,
title={A Fast and Scalable Joint Estimator for Learning Multiple Related Sparse Gaussian Graphical Models},
author={Wang, Beilun and Gao, Ji and Qi, Yanjun},
booktitle={Proceedings of the 20th International Conference on Artificial Intelligence and Statistics, PMLR:, 2017.},
volume={54},
pages={1168--1177},
year={2017}
}
Having trouble with our tools? Please contact Beilun and we’ll help you sort it out.
08 May 2016
install.packages("simule")
library(simule)
demo(simule)
Abstract
Identifying context-specific entity networks from aggregated data is an important task, arising often in bioinformatics and neuroimaging. Computationally, this task can be formulated as jointly estimating multiple different, but related, sparse Undirected Graphical Models (UGM) from aggregated samples across several contexts. Previous joint-UGM studies have mostly focused on sparse Gaussian Graphical Models (sGGMs) and can’t identify context-specific edge patterns directly. We, therefore, propose a novel approach, SIMULE (detecting Shared and Individual parts of MULtiple graphs Explicitly) to learn multi-UGM via a constrained L1 minimization. SIMULE automatically infers both specific edge patterns that are unique to each context and shared interactions preserved among all the contexts. Through the L1 constrained formulation, this problem is cast as multiple independent subtasks of linear programming that can be solved efficiently in parallel. In addition to Gaussian data, SIMULE can also handle multivariate Nonparanormal data that greatly relaxes the normality assumption that many real-world applications do not follow. We provide a novel theoretical proof showing that SIMULE achieves a consistent result at the rate O(log(Kp)/n_{tot}). On multiple synthetic datasets and two biomedical datasets, SIMULE shows significant improvement over state-of-the-art multi-sGGM and single-UGM baselines.
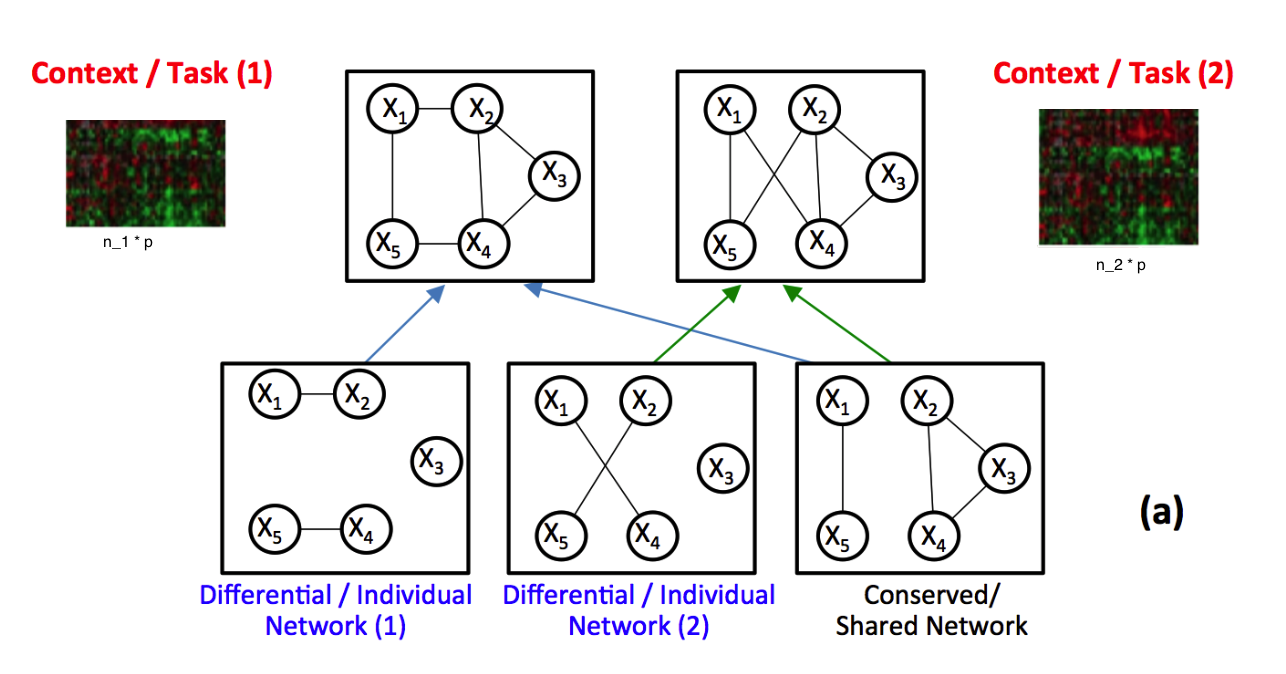
Citations
@Article{Wang2017,
author="Wang, Beilun and Singh, Ritambhara and Qi, Yanjun",
title="A constrained L1 minimization approach for estimating multiple sparse Gaussian or nonparanormal graphical models",
journal="Machine Learning",
year="2017",
month="Oct",
day="01",
volume="106",
number="9",
pages="1381--1417",
abstract="Identifying context-specific entity networks from aggregated data is an important task, arising often in bioinformatics and neuroimaging applications. Computationally, this task can be formulated as jointly estimating multiple different, but related, sparse undirected graphical models(UGM) from aggregated samples across several contexts. Previous joint-UGM studies have mostly focused on sparse Gaussian graphical models (sGGMs) and can't identify context-specific edge patterns directly. We, therefore, propose a novel approach, SIMULE (detecting Shared and Individual parts of MULtiple graphs Explicitly) to learn multi-UGM via a constrained L1 minimization. SIMULE automatically infers both specific edge patterns that are unique to each context and shared interactions preserved among all the contexts. Through the L1 constrained formulation, this problem is cast as multiple independent subtasks of linear programming that can be solved efficiently in parallel. In addition to Gaussian data, SIMULE can also handle multivariate Nonparanormal data that greatly relaxes the normality assumption that many real-world applications do not follow. We provide a novel theoretical proof showing that SIMULE achieves a consistent result at the rate
log (Kp)/(n_tot). On multiple synthetic datasets and two biomedical datasets, SIMULE shows significant improvement over state-of-the-art multi-sGGM and single-UGM baselines
(SIMULE implementation and the used datasets @ https://github.com/QData/SIMULE ).",
issn="1573-0565",
doi="10.1007/s10994-017-5635-7",
url="https://doi.org/10.1007/s10994-017-5635-7"
}
Having trouble with our tools? Please contact Beilun and we’ll help you sort it out.
11 Jun 2015
Abstract
Predicting protein properties such as solvent accessibility and secondary structure from its primary amino acid sequence is an important task in bioinformatics. Recently, a few deep learning models have surpassed the traditional window based multilayer perceptron. Taking inspiration from the image classification domain we propose a deep convolutional neural network architecture, MUST-CNN, to predict protein properties. This architecture uses a novel multilayer shift-and-stitch (MUST) technique to generate fully dense per-position predictions on protein sequences. Our model is significantly simpler than the state-of-the-art, yet achieves better results. By combining MUST and the efficient convolution operation, we can consider far more parameters while retaining very fast prediction speeds. We beat the state-of-the-art performance on two large protein property prediction datasets.
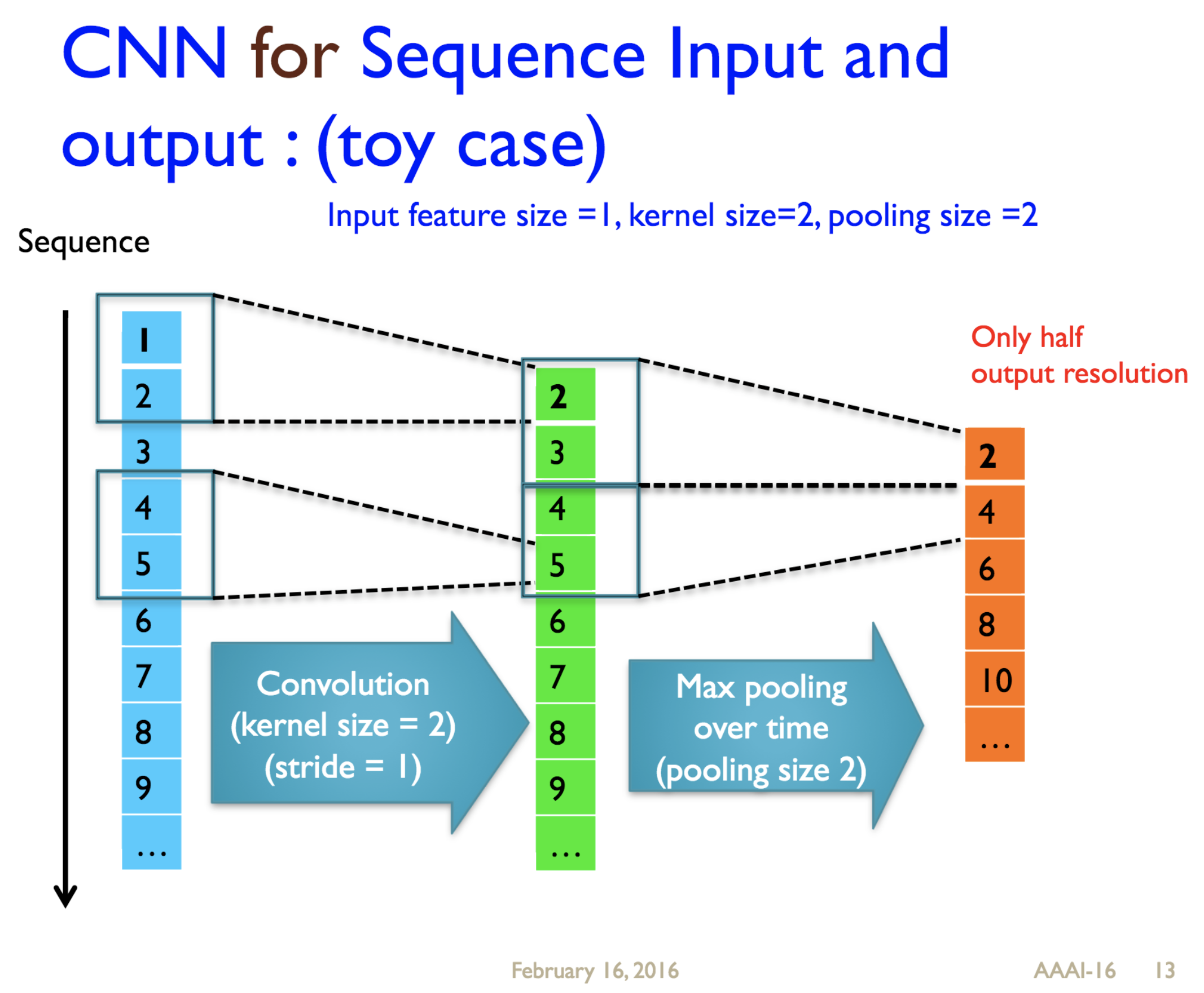
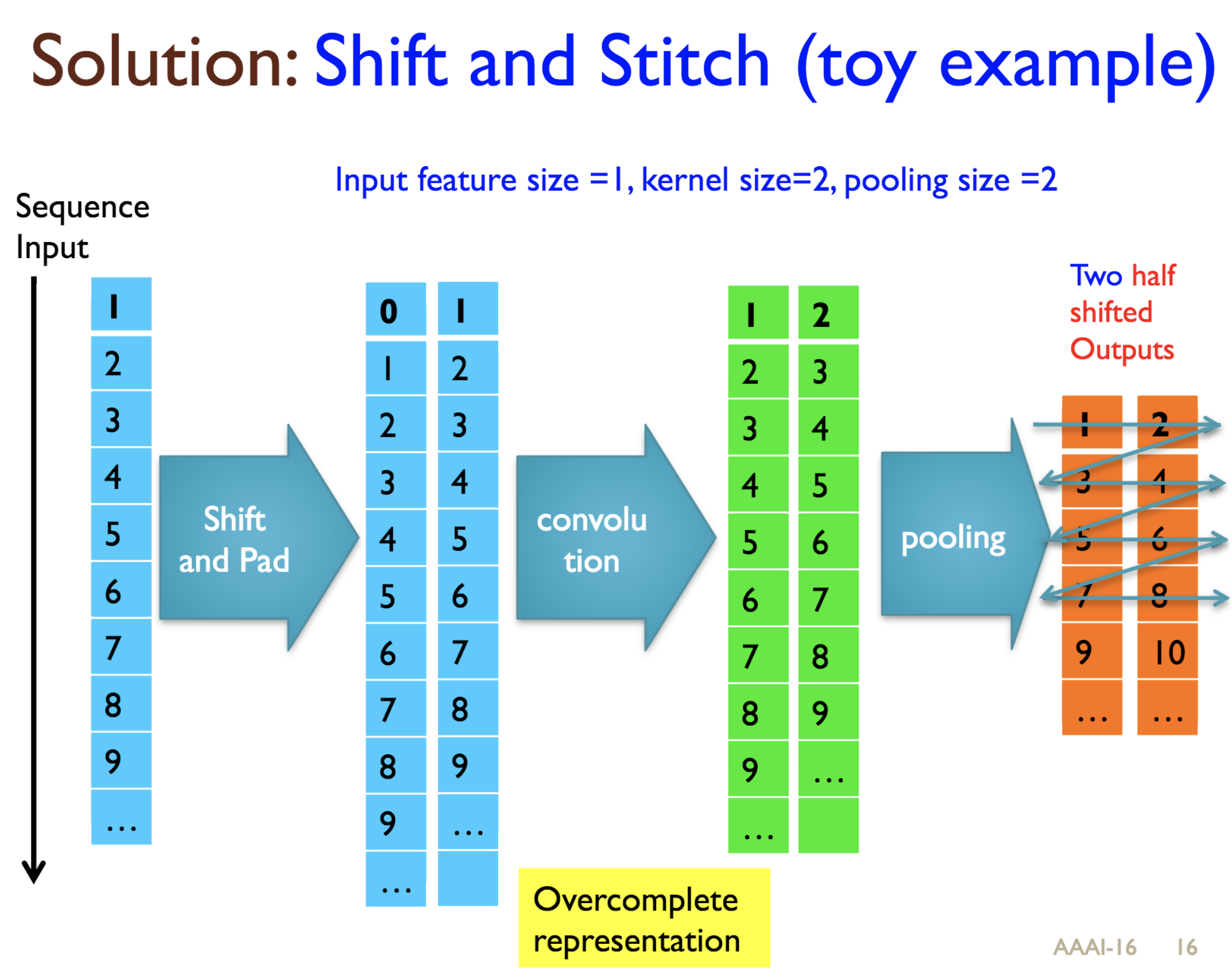
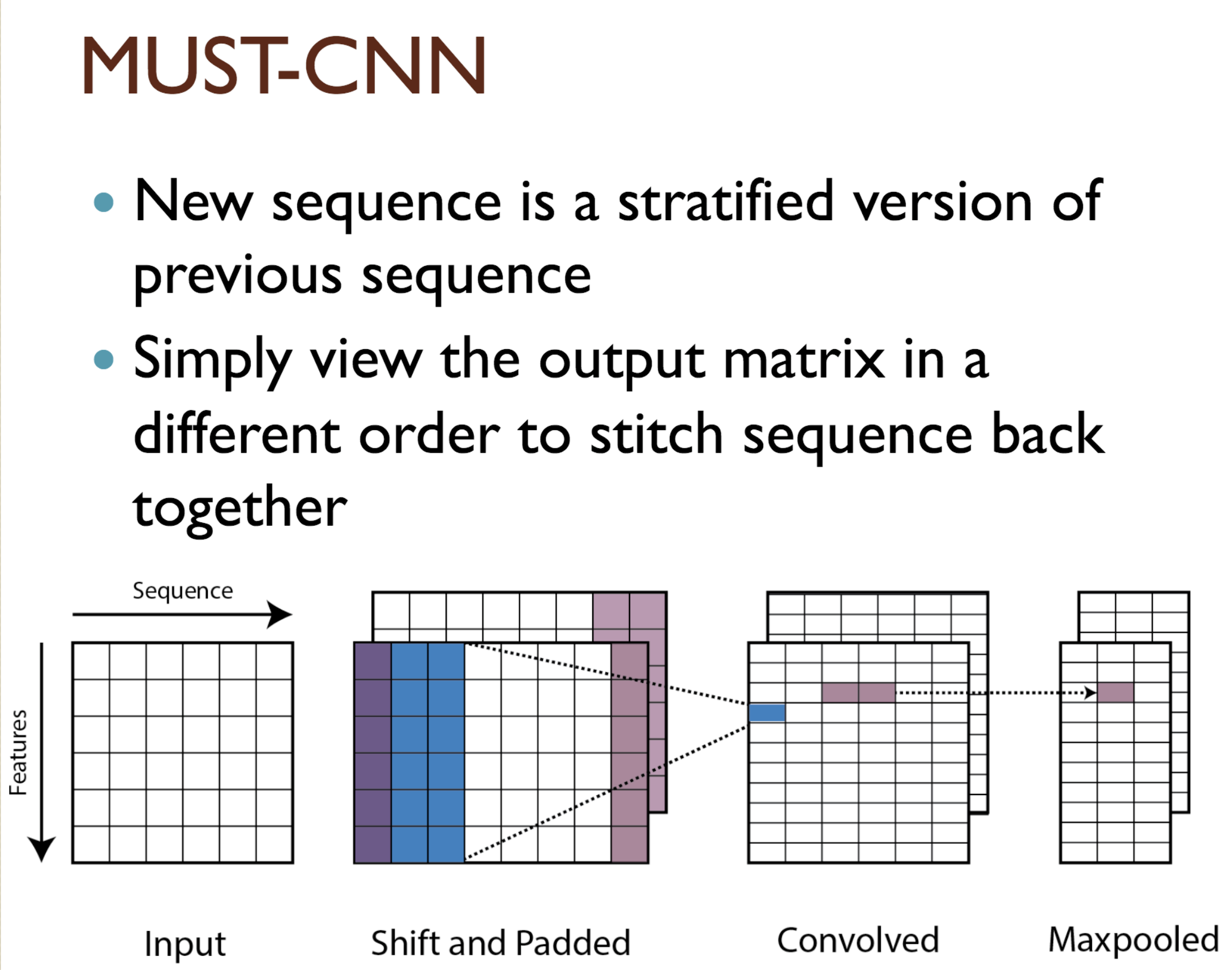

Citations
@inproceedings{lin2016must,
title={MUST-CNN: a multilayer shift-and-stitch deep convolutional architecture for sequence-based protein structure prediction},
author={Lin, Zeming and Lanchantin, Jack and Qi, Yanjun},
booktitle={Proceedings of the Thirtieth AAAI Conference on Artificial Intelligence},
pages={27--34},
year={2016},
organization={AAAI Press}
}
Having trouble with our tools? Please contact Jack and we’ll help you sort it out.
20 Jun 2014
- Yunlong He, Yanjun Qi, Koray Kavukcuoglu, Haesun Park
Abstract:
In this paper, we study latent factor models with the dependency structure in the latent space. We propose a general learning framework which induces sparsity on the undirected graphical model imposed on the vector of latent factors. A novel latent factor model SLFA is then proposed as a matrix factorization problem with a special regularization term that encourages collaborative reconstruction. The main benefit (novelty) of the model is that we can simultaneously learn the lower-dimensional representation for data and model the pairwise relationships between latent factors explicitly. An on-line learning algorithm is devised to make the model feasible for large-scale learning problems. Experimental results on two synthetic data and two real-world data sets demonstrate that pairwise relationships and latent factors learned by our model provide a more structured way of exploring high-dimensional data, and the learned representations achieve the state-of-the-art classification performance.
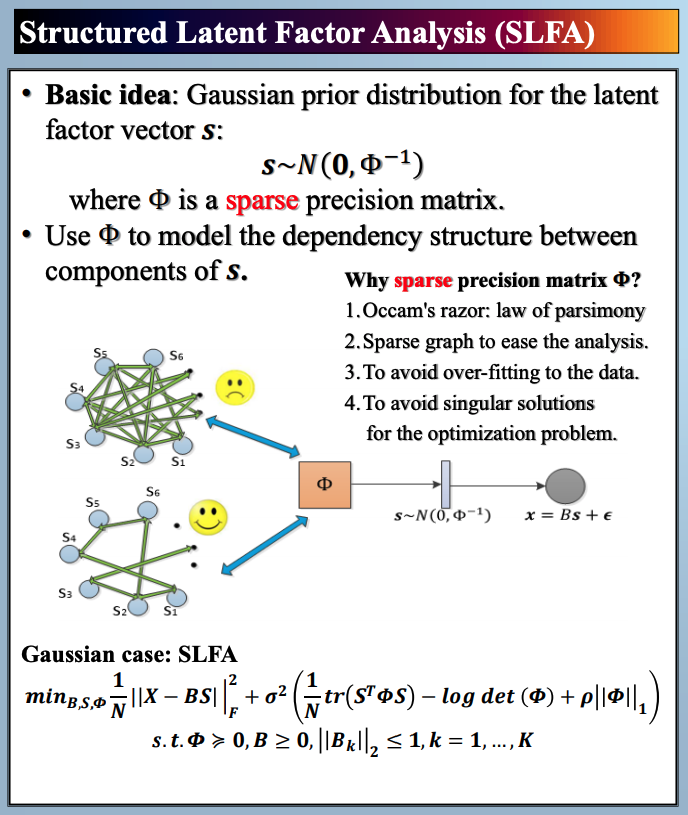
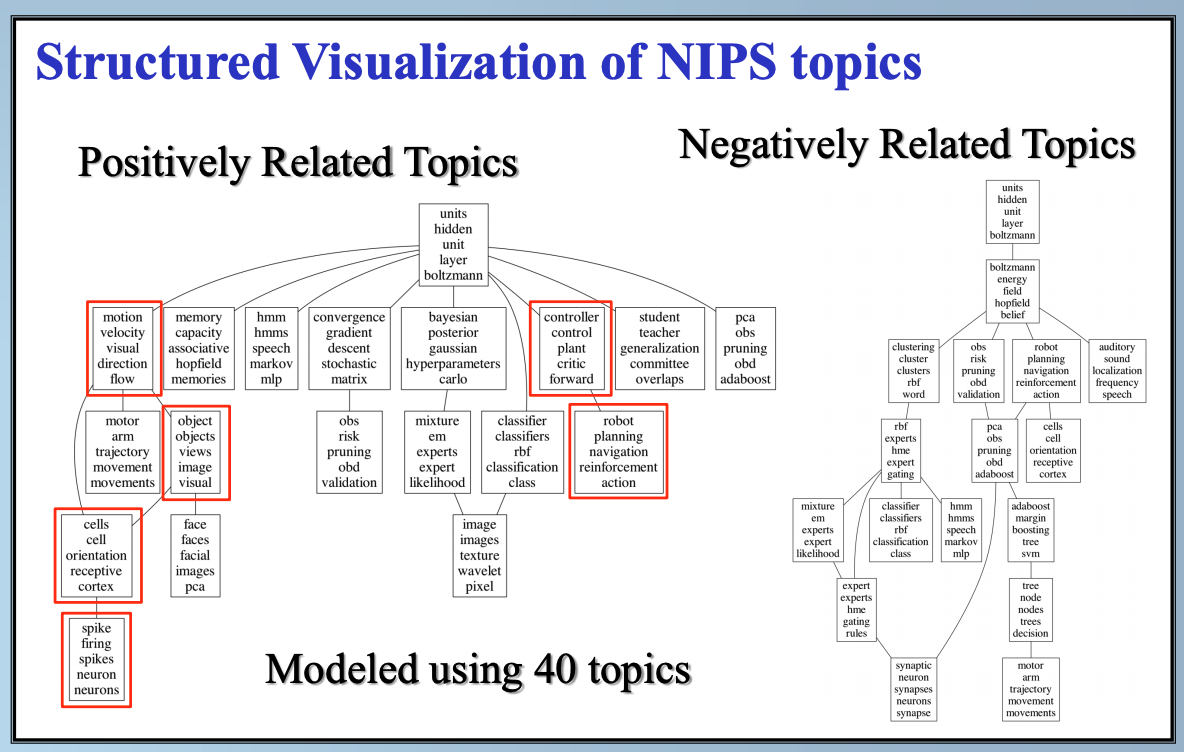

Citations
@inproceedings{he2012learning,
title={Learning the dependency structure of latent factors},
author={He, Yunlong and Qi, Yanjun and Kavukcuoglu, Koray and Park, Haesun},
booktitle={Advances in neural information processing systems},
pages={2366--2374},
year={2012}
}
Having trouble with our tools? Please contact Yanjun Qi and we’ll help you sort it out.
20 Jun 2014
- Y He, K Kavukcuoglu, Y Wang, A Szlam, Y Qi
Abstract:
In this paper, we propose a new unsupervised feature learning framework, namely Deep Sparse Coding (DeepSC), that extends sparse coding to a multi-layer architecture for visual object recognition tasks. The main innovation of the framework is that it connects the sparse-encoders from different layers by a sparse-to-dense module. The sparse-to-dense module is a composition of a local spatial pooling step and a low-dimensional embedding process, which takes advantage of the spatial smoothness information in the image. As a result, the new method is able to learn several levels of sparse representation of the image which capture features at a variety of abstraction levels and simultaneously preserve the spatial smoothness between the neighboring image patches. Combining the feature representations from multiple layers, DeepSC achieves the state-of-the-art performance on multiple object recognition tasks.
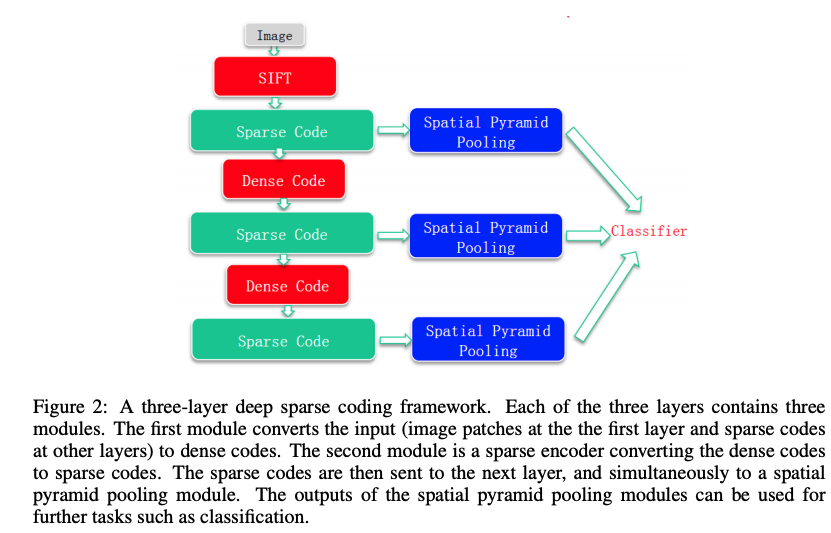
Citations
@misc{he2013unsupervised,
title={Unsupervised Feature Learning by Deep Sparse Coding},
author={Yunlong He and Koray Kavukcuoglu and Yun Wang and Arthur Szlam and Yanjun Qi},
year={2013},
eprint={1312.5783},
archivePrefix={arXiv},
primaryClass={cs.LG}
}
Having trouble with our tools? Please contact Yanjun Qi and we’ll help you sort it out.
01 Oct 2013
- authors: Yanjun Qi, Sujatha Das, Ronan Collobert, Jason Weston
Supplementary Here
Abstract
In this paper we introduce a deep neural network architecture to perform information extraction on character-based sequences,
e.g. named-entity recognition on Chinese text or secondary-structure detection on protein sequences. With a task-independent architecture, the
deep network relies only on simple character-based features, which obviates the need for task-specific feature engineering. The proposed discriminative framework includes three important strategies, (1) a deep
learning module mapping characters to vector representations is included
to capture the semantic relationship between characters; (2) abundant
online sequences (unlabeled) are utilized to improve the vector representation through semi-supervised learning; and (3) the constraints of
spatial dependency among output labels are modeled explicitly in the
deep architecture. The experiments on four benchmark datasets have
demonstrated that, the proposed architecture consistently leads to the
state-of-the-art performance.
Citations
@inproceedings{qi2014deep,
title={Deep learning for character-based information extraction},
author={Qi, Yanjun and Das, Sujatha G and Collobert, Ronan and Weston, Jason},
booktitle={European Conference on Information Retrieval},
pages={668--674},
year={2014},
organization={Springer}
}
Having trouble with our tools? Please contact Yanjun Qi and we’ll help you sort it out.
08 Mar 2013
Here are the slides of one lecture talk I gave at UVA CPHG Seminar Series in 2014 about our deep learning tools back then.
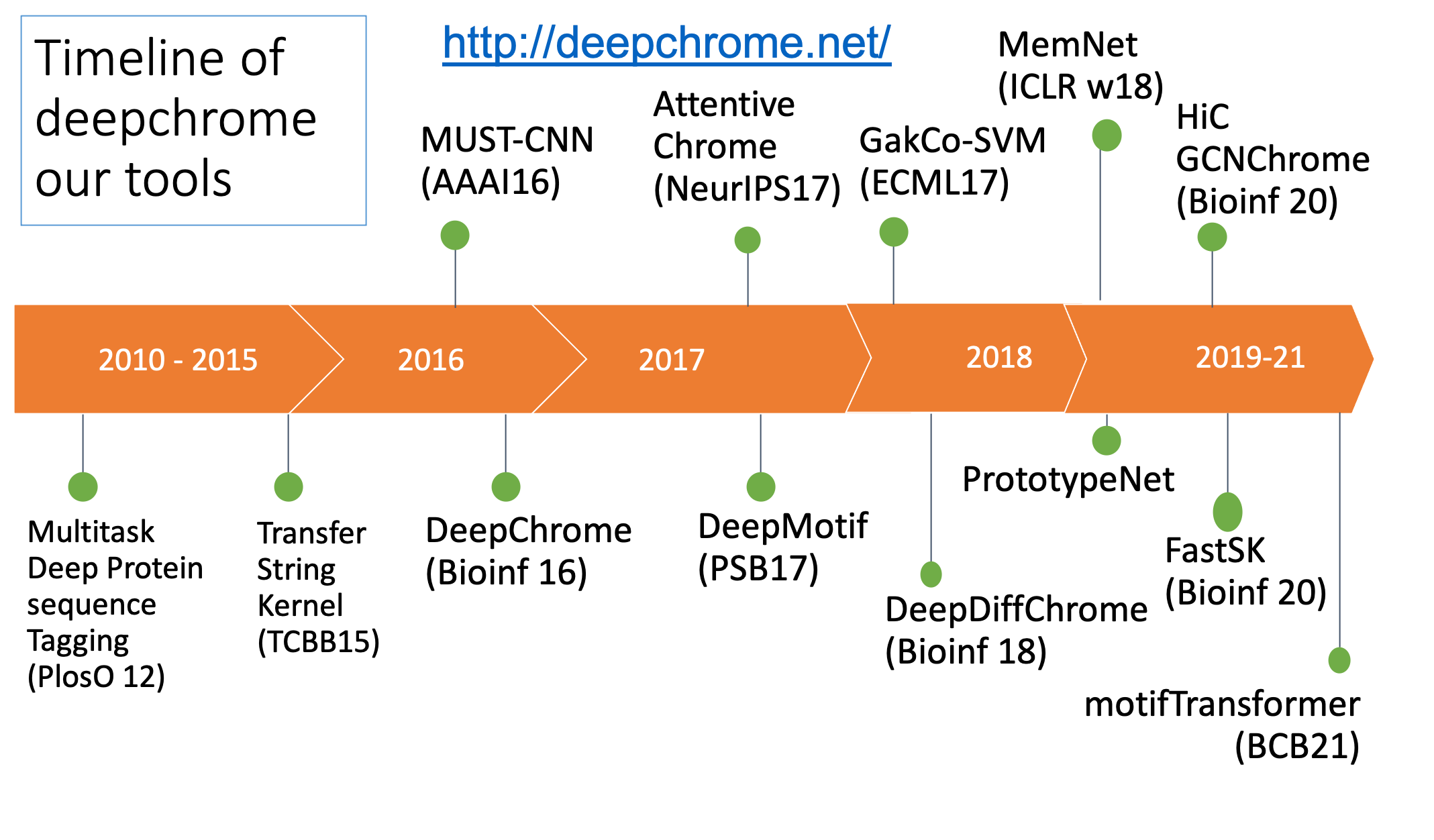
Thanks for reading!
12 Jan 2013
Abstract
A variety of functionally important protein properties, such as secondary structure, transmembrane topology and solvent accessibility, can be encoded as a labeling of amino acids. Indeed, the prediction of such properties from the primary amino acid sequence is one of the core projects of computational biology. Accordingly, a panoply of approaches have been developed for predicting such properties; however, most such approaches focus on solving a single task at a time. Motivated by recent, successful work in natural language processing, we propose to use multitask learning to train a single, joint model that exploits the dependencies among these various labeling tasks. We describe a deep neural network architecture that, given a protein sequence, outputs a host of predicted local properties, including secondary structure, solvent accessibility, transmembrane topology, signal peptides and DNA-binding residues. The network is trained jointly on all these tasks in a supervised fashion, augmented with a novel form of semi-supervised learning in which the model is trained to distinguish between local patterns from natural and synthetic protein sequences. The task-independent architecture of the network obviates the need for task-specific feature engineering. We demonstrate that, for all of the tasks that we considered, our approach leads to statistically significant improvements in performance, relative to a single task neural network approach, and that the resulting model achieves state-of-the-art performance.
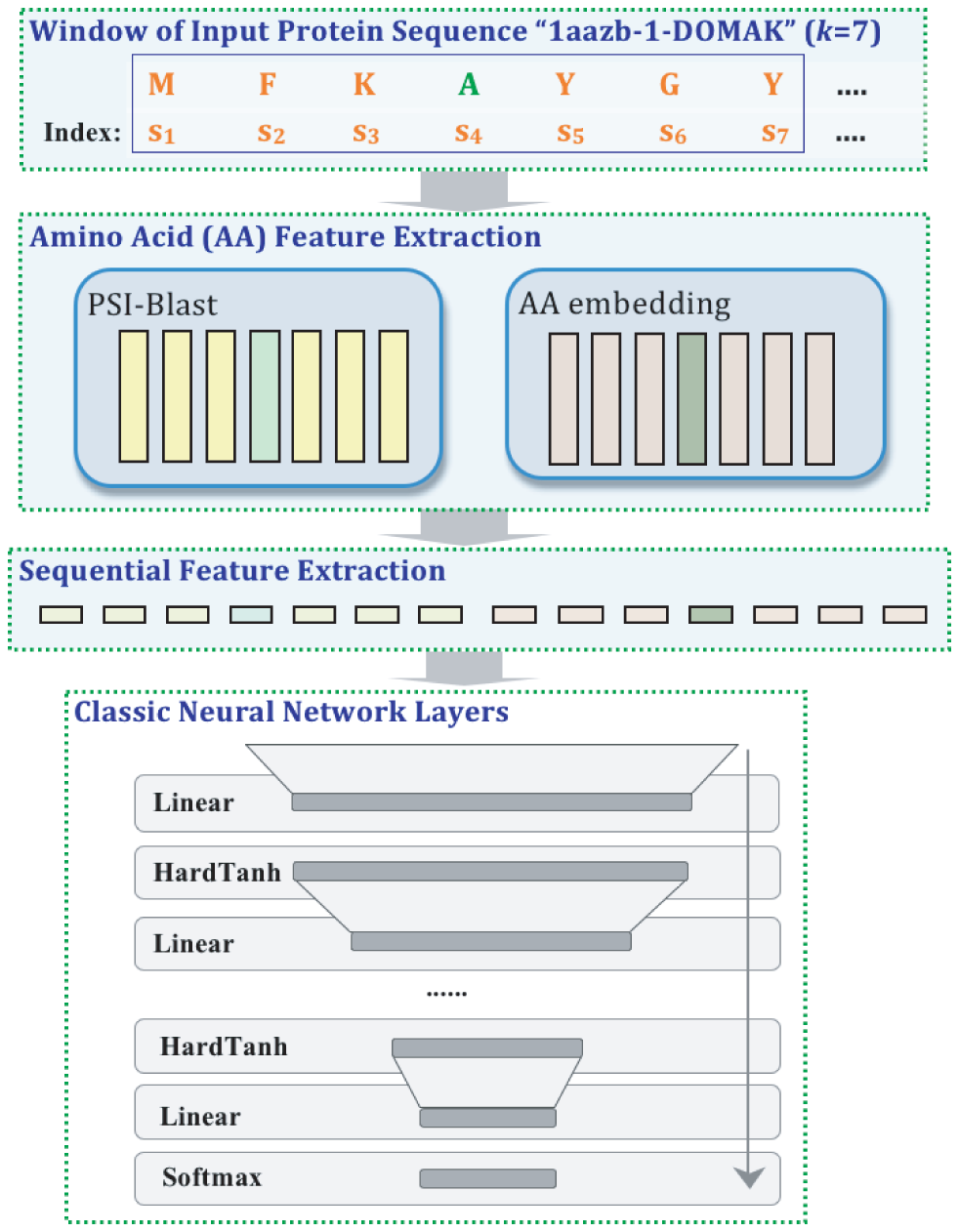
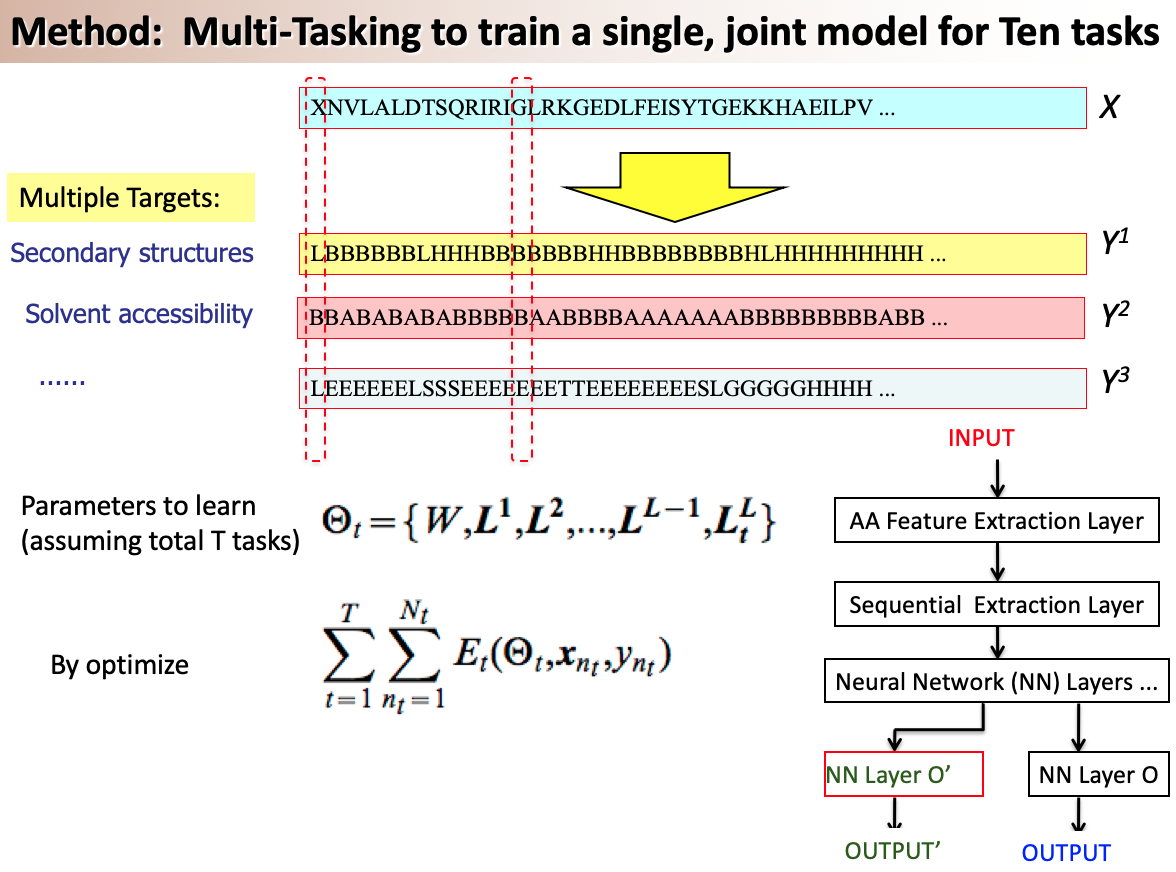
Citations
@article{qi12plosone,
author = {Qi, , Yanjun AND Oja, , Merja AND Weston, , Jason AND Noble, , William Stafford},
journal = {PLoS ONE},
publisher = {Public Library of Science},
title = {A Unified Multitask Architecture for Predicting Local Protein Properties},
year = {2012},
month = {03},
volume = {7},
url = {http://dx.doi.org/10.1371%2Fjournal.pone.0032235},
pages = {e32235},
number = {3},
doi = {10.1371/journal.pone.0032235}
}
Having trouble with our tools? Please contact Jack and we’ll help you sort it out.
01 May 2012
Paper1: Learning the Dependency Structure of Latent Factors
- Y. He, Y. Qi, K. Kavukcuoglu, H. Park (2012) NeurIPS
- PDF
-
Talk: Slide
- Abstract
In this paper, we study latent factor models with the dependency structure in the latent space. We propose a general learning framework which induces sparsity on the undirected graphical model imposed on the vector of latent factors. A novel latent factor model SLFA is then proposed as a matrix factorization problem with a special regularization term that encourages collaborative reconstruction. The main benefit (novelty) of the model is that we can simultaneously learn the lower-dimensional representation for data and model the pairwise relationships between latent factors explicitly. An on-line learning algorithm is devised to make the model feasible for large-scale learning problems. Experimental results on two synthetic data and two real-world data sets demonstrate that pairwise relationships and latent factors learned by our model provide a more structured way of exploring high-dimensional data, and the learned representations achieve the state-of-the-art classification performance.
Citations
@INPROCEEDINGS{yhe12NIPS,
title={Learning the Dependency Structure of Latent Factors},
author={Y. He and Y. Qi and K. Kavukcuoglu and H. Park},
booktitle={Proceedings of Advances in Neural Information Processing Systems (NIPS)},
year={2012},
note="{\\Acceptance rate = 25\% (370/1467)}"
}
Paper2: Sparse higher-order Markov random field
-
PDF
-
Abstract
Systems and methods are provided for identifying combinatorial feature interactions, including capturing statistical dependencies between categorical variables, with the statistical dependencies being stored in a computer readable storage medium. A model is selected based on the statistical dependencies using a neighborhood estimation strategy, with the neighborhood estimation strategy including generating sets of arbitrarily high-order feature interactions using at least one rule forest and optimizing one or more likelihood functions. A damped mean-field approach is applied to the model to obtain parameters of a Markov random field (MRF); a sparse high-order semi-restricted MRF is produced by adding a hidden layer to the MRF; indirect long-range dependencies between feature groups are modeled using the sparse high-order semi-restricted MRF; and a combinatorial dependency structure between variables is output.
Citations
@misc{min2015sparse,
title={Sparse higher-order Markov random field},
author={Min, Renqiang and Qi, Yanjun},
year={2015},
month=nov # "~10",
publisher={Google Patents},
note={US Patent 9,183,503}
}
Having trouble with our tools? Please contact Yanjun Qi and we’ll help you sort it out.
01 May 2011
Paper0: Learning to rank with (a lot of) word features
-
PDF
-
Abstract
In this article we present Supervised Semantic Indexing which defines a class of nonlinear (quadratic) models that are discriminatively trained to directly map from the word content in a query-document or document-document pair to a ranking score. Like Latent Semantic Indexing (LSI), our models take account of correlations between words (synonymy, polysemy). However, unlike LSI our models are trained from a supervised signal directly on the ranking task of interest, which we argue is the reason for our superior results. As the query and target texts are modeled separately, our approach is easily generalized to different retrieval tasks, such as cross-language retrieval or online advertising placement. Dealing with models on all pairs of words features is computationally challenging. We propose several improvements to our basic model for addressing this issue, including low rank (but diagonal preserving) representations, correlated feature hashing and sparsification. We provide an empirical study of all these methods on retrieval tasks based on Wikipedia documents as well as an Internet advertisement task. We obtain state-of-the-art performance while providing realistically scalable methods.
Paper1: Polynomial semantic indexing
- PDF
- Bing Bai, Jason Weston, David Grangier, Ronan Collobert, Kunihiko Sadamasa, Yanjun Qi, Corinna Cortes, Mehryar Mohri
- 2009 Conference on Advances in Neural Information Processing Systems
- Abstract
We present a class of nonlinear (polynomial) models that are discriminatively trained to directly map from the word content in a query-document or document-document pair to a ranking score. Dealing with polynomial models on word features is computationally challenging. We propose a low rank (but diagonal preserving) representation of our polynomial models to induce feasible memory and computation requirements. We provide an empirical study on retrieval tasks based on Wikipedia documents, where we obtain state-of-the-art performance while providing realistically scalable methods.
Paper2: Retrieving Medical Records with “sennamed”: NEC Labs America at TREC 2012 Medical Records Track
-
PDF
-
Abstract
In this notebook, we describe the automatic retrieval runs from NEC Laboratories America (NECLA) for the Text REtrieval Conference (TREC) 2012 Medical Records track. Our approach is based on a combination of UMLS medical concept detection and a set of simple retrieval models. Our best run, sennamed2, has achieved the best inferred average precision (infAP) score on 5 of the 47 test topics, and obtained a higher score than the median of all submission runs on 27 other topics. Overall, sennamed2 ranks at the second place amongst all the 82 automatic runs submitted for this track, and obtains the third place amongst both automatic and manual submissions.
-
PDF
-
Abstract
With the advancement of genome-wide monitoring technologies, molecular expression data have become widely used for diagnosing cancer through tumor or blood samples. When mining molecular signature data, the process of comparing samples through an adaptive distance function is fundamental but difficult, as such datasets are normally heterogeneous and high dimensional. In this article, we present kernelized information-theoretic metric learning (KITML) algorithms that optimize a distance function to tackle the cancer diagnosis problem and scale to high dimensionality. By learning a nonlinear transformation in the input space implicitly through kernelization, KITML permits efficient optimization, low storage, and improved learning of distance metric. We propose two novel applications of KITML for diagnosing cancer using high-dimensional molecular profiling data: (1) for sample-level cancer diagnosis, the learned metric is used to improve the performance of k-nearest neighbor classification; and (2) for estimating the severity level or stage of a group of samples, we propose a novel set-based ranking approach to extend KITML. For the sample-level cancer classification task, we have evaluated on 14 cancer gene microarray datasets and compared with eight other state-of-the-art approaches. The results show that our approach achieves the best overall performance for the task of molecular-expression-driven cancer sample diagnosis. For the group-level cancer stage estimation, we test the proposed set-KITML approach using three multi-stage cancer microarray datasets, and correctly estimated the stages of sample groups for all three studies.
Paper4: Learning preferences with millions of parameters by enforcing sparsity
- PDF
-
Talk
- Abstract
We study the retrieval task that ranks a set of objects for a given query in the pair wise preference learning framework. Recently researchers found out that raw features (e.g. words for text retrieval) and their pair wise features which describe relationships between two raw features (e.g. word synonymy or polysemy) could greatly improve the retrieval precision. However, most existing methods can not scale up to problems with many raw features (e.g. English vocabulary), due to the prohibitive computational cost on learning and the memory requirement to store a quadratic number of parameters. In this paper, we propose to learn a sparse representation of the pair wise features under the preference learning framework using the L1 regularization. Based on stochastic gradient descent, an online algorithm is devised to enforce the sparsity using a mini-batch shrinkage strategy. On multiple benchmark datasets, we show that our method achieves better performance with fast convergence, and takes much less memory on models with millions of parameters.
Citations
@techreport{qi2012retrieving,
title={Retrieving medical records with sennamed: Nec labs america at trec 2012 medical records track},
author={Qi, Yanjun and Laquerre, Pierre-Fran{\c{c}}ois},
year={2012},
institution={NEC Laboratories America Inc Princeton NJ}
}
Having trouble with our tools? Please contact Yanjun Qi and we’ll help you sort it out.
01 May 2011
Summary:
-
Methods and systems for document classification include embedding n-grams from an input text in a latent space, embedding the input text in the latent space based on the embedded n-grams and weighting said n-grams according to spatial evidence of the respective n-grams in the input text, classifying the document along one or more axes, and adjusting weights used to weight the n-grams based on the output of the classifying step.
-
authors: Qi, Yanjun and Bai, Bing
Paper1: Sentiment classification with supervised sequence embedding
- PDF
-
Talk: Slide
- Abstract
In this paper, we introduce a novel approach for modeling n-grams in a latent space learned from supervised signals. The proposed procedure uses only unigram features to model short phrases (n-grams) in the latent space. The phrases are then combined to form document-level latent representation for a given text, where position of an n-gram in the document is used to compute corresponding combining weight. The resulting two-stage supervised embedding is then coupled with a classifier to form an end-to-end system that we apply to the large-scale sentiment classification task. The proposed model does not require feature selection to retain effective features during pre-processing, and its parameter space grows linearly with size of n-gram. We present comparative evaluations of this method using two large-scale datasets for sentiment classification in online reviews (Amazon and TripAdvisor). The proposed method outperforms standard baselines that rely on bag-of-words representation populated with n-gram features.
Paper2: Sentiment Classification Based on Supervised Latent n-gram Analysis
- PDF
-
Talk: Slide
- Abstract
In this paper, we propose an efficient embedding for modeling higher-order (n-gram) phrases that projects the n-grams to low-dimensional latent semantic space, where a classification function can be defined. We utilize a deep neural network to build a unified discriminative framework that allows for estimating the parameters of the latent space as well as the classification function with a bias for the target classification task at hand. We apply the framework to large-scale sentimental classification task. We present comparative evaluation of the proposed method on two (large) benchmark data sets for online product reviews. The proposed method achieves superior performance in comparison to the state of the art.
Citations
@misc{qi2014document,
title={Document classification with weighted supervised n-gram embedding},
author={Qi, Yanjun and Bai, Bing},
year={2014},
month=nov # "~18",
publisher={Google Patents},
note={US Patent 8,892,488}
}
Having trouble with our tools? Please contact Yanjun Qi and we’ll help you sort it out.
01 Mar 2010
Title: Systems and methods for semi-supervised relationship extraction
- authors: Qi, Yanjun and Bai, Bing and Ning, Xia and Kuksa, Pavel
- PDF
-
Talk: Slide
- Abstract
Bio-relation extraction (bRE), an important goal in bio-text mining, involves subtasks identifying relationships between bio-entities in text at multiple levels, e.g., at the article, sentence or relation level. A key limitation of current bRE systems is that they are restricted by the availability of annotated corpora. In this work we introduce a semi-supervised approach that can tackle multi-level bRE via string comparisons with mismatches in the string kernel framework. Our string kernel implements an abstraction step, which groups similar words to generate more abstract entities, which can be learnt with unlabeled data. Specifically, two unsupervised models are proposed to capture contextual (local or global) semantic similarities between words from a large unannotated corpus. This Abstraction-augmented String Kernel (ASK) allows for better generalization of patterns learned from annotated data and provides a unified framework for solving bRE with multiple degrees
of detail. ASK shows effective improvements over classic string kernels
on four datasets and achieves state-of-the-art bRE performance without
the need for complex linguistic features.
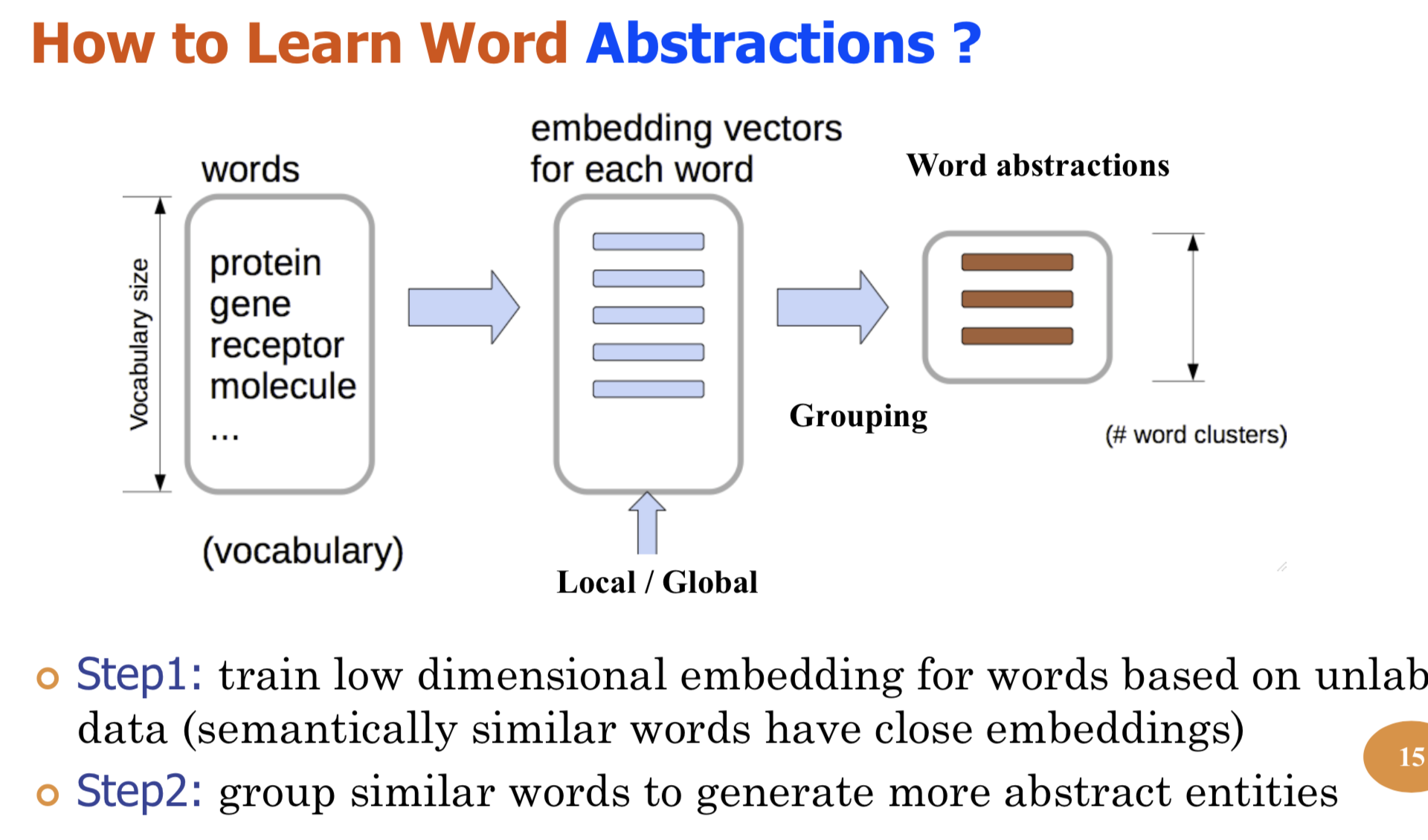
-
PDF
- Talk: Slide
-
URL More
- Abstract
Extracting semantic relations between entities is an important step towards automatic text understanding. In this paper, we propose a novel Semi-supervised Convolution Graph Kernel (SCGK) method for semantic Relation Extraction (RE) from natural language. By encoding English sentences as dependence graphs among words, SCGK computes kernels (similarities) between sentences using a convolution strategy, i.e., calculating similarities over all possible short single paths from two dependence graphs. Furthermore, SCGK adds three semi-supervised strategies in the kernel calculation to incorporate soft-matches between (1) words, (2) grammatical dependencies, and (3) entire sentences, respectively. From a large unannotated corpus, these semi-supervision steps learn to capture contextual semantic patterns of elements in natural sentences, which therefore alleviate the lack of annotated examples in most RE corpora. Through convolutions and multi-level semi-supervisions, SCGK provides a powerful model to encode both syntactic and semantic evidence existing in natural English sentences, which effectively recovers the target relational patterns of interest. We perform extensive experiments on five RE benchmark datasets which aim to identify interaction relations from biomedical literature. Our results demonstrate that SCGK achieves the state-of-the-art performance on the task of semantic relation extraction.
Paper3: Semi-Supervised Bio-Named Entity Recognition with Word-Codebook Learning
- Pavel P. Kuksa, Yanjun Qi,
-
PDF
- Abstract
We describe a novel semi-supervised method called WordCodebook Learning (WCL), and apply it to the task of bionamed entity recognition (bioNER). Typical bioNER systems can be seen as tasks of assigning labels to words in bioliterature text. To improve supervised tagging, WCL learns
a class of word-level feature embeddings to capture word
semantic meanings or word label patterns from a large unlabeled corpus. Words are then clustered according to their
embedding vectors through a vector quantization step, where
each word is assigned into one of the codewords in a codebook. Finally codewords are treated as new word attributes
and are added for entity labeling. Two types of wordcodebook learning are proposed: (1) General WCL, where
an unsupervised method uses contextual semantic similarity of words to learn accurate word representations; (2)
Task-oriented WCL, where for every word a semi-supervised
method learns target-class label patterns from unlabeled
data using supervised signals from trained bioNER model.
Without the need for complex linguistic features, we demonstrate utility of WCL on the BioCreativeII gene name recognition competition data, where WCL yields state-of-the-art
performance and shows great improvements over supervised
baselines and semi-supervised counter peers.
Citations
@INPROCEEDINGS{ecml2010ask,
author = {Pavel P. Kuksa and Yanjun Qi and Bing Bai and Ronan Collobert and
Jason Weston and Vladimir Pavlovic and Xia Ning},
title = {Semi-Supervised Abstraction-Augmented String Kernel for Multi-Level
Bio-Relation Extraction},
booktitle = {ECML},
year = {2010},
note = {Acceptance rate: 106/658 (16%)},
bib2html_pubtype = {Refereed Conference},
}
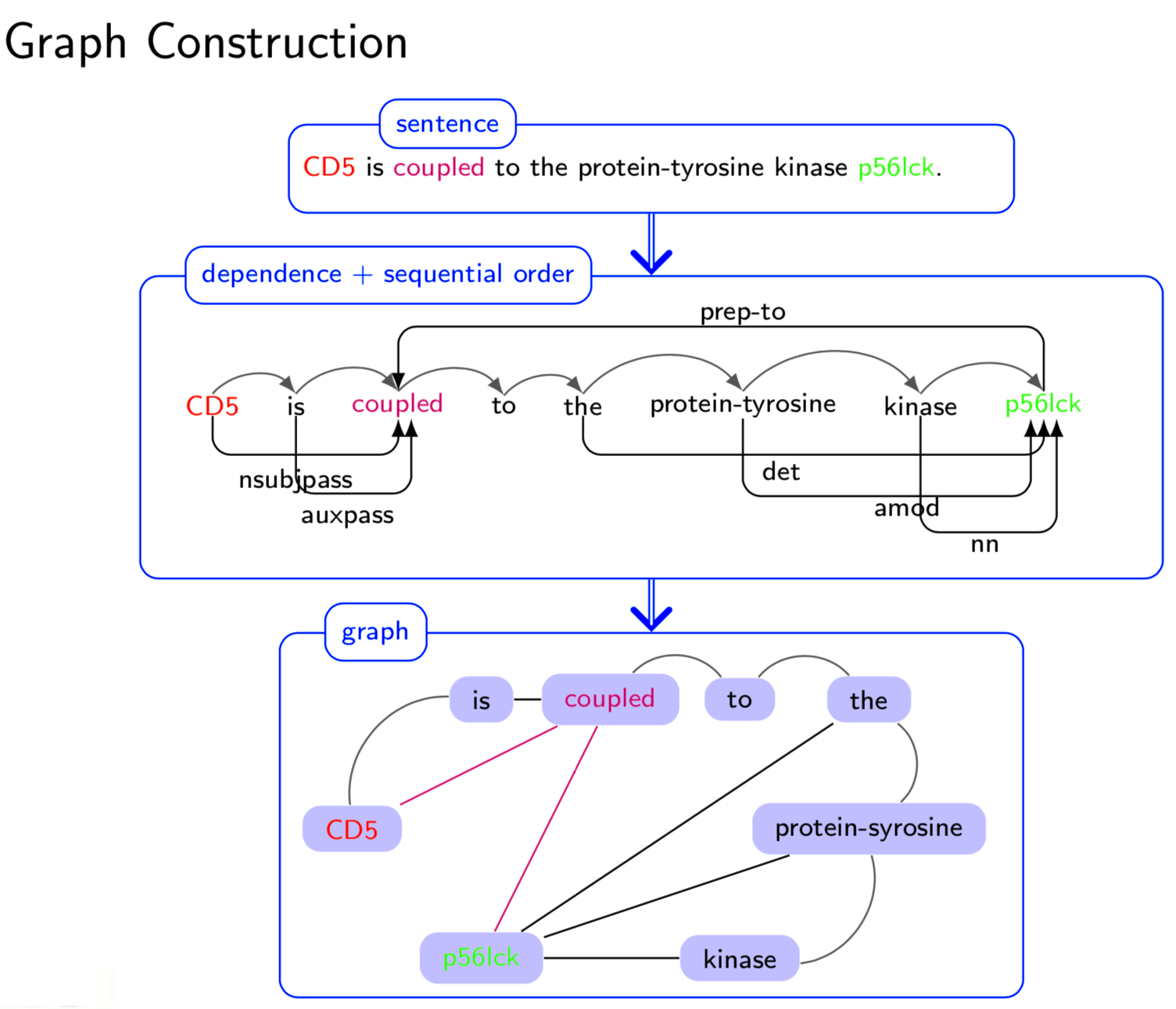
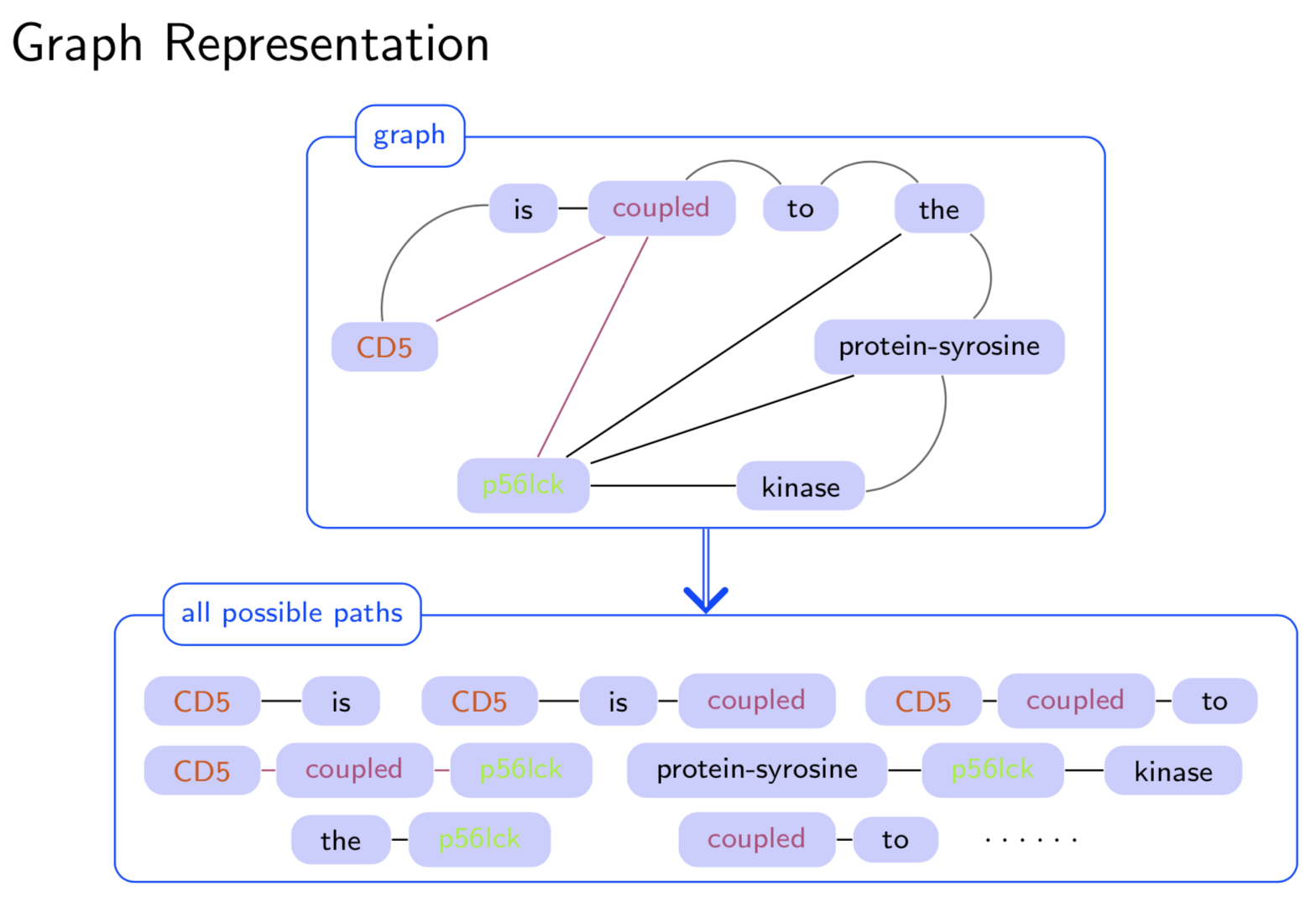

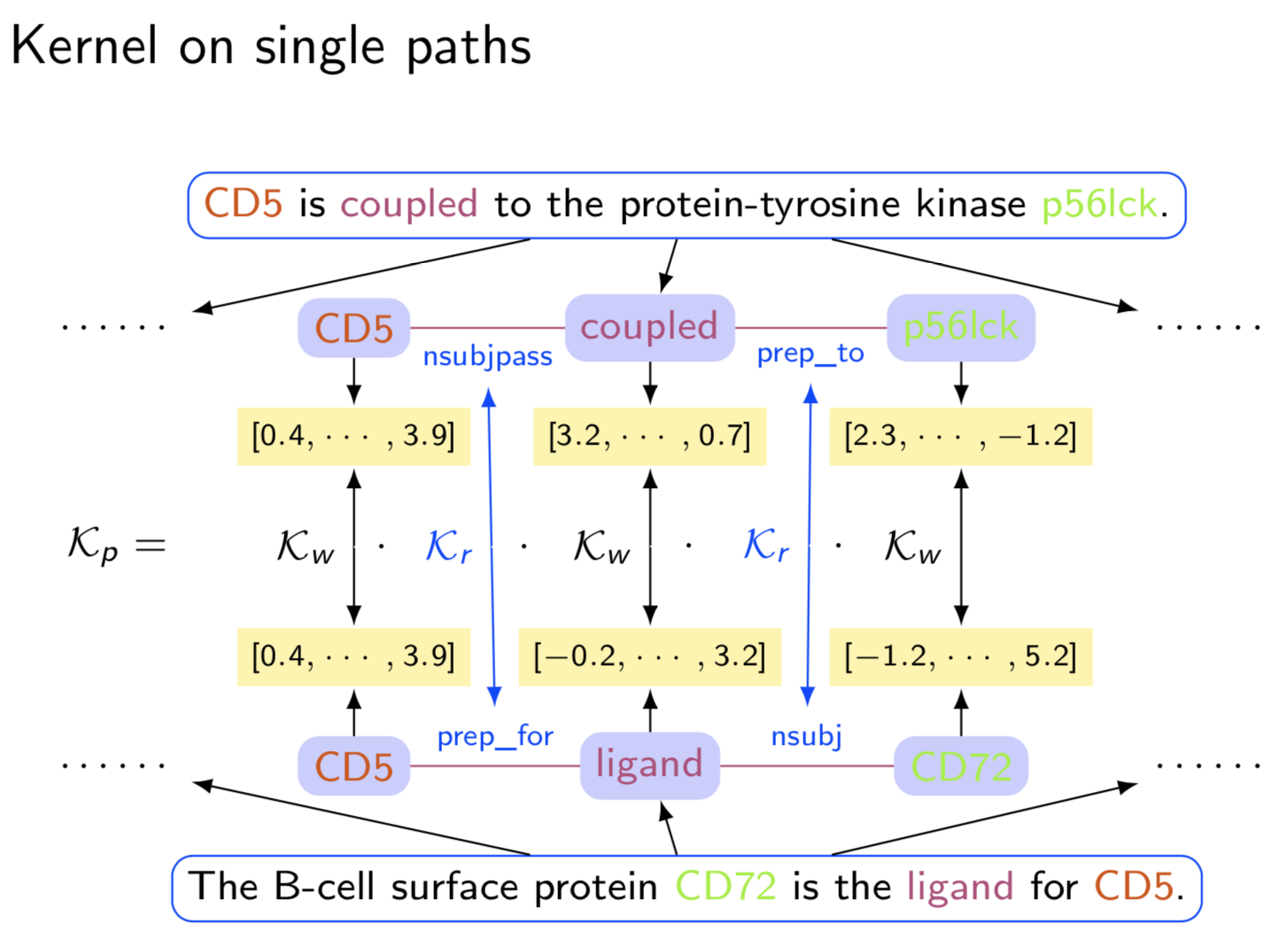

Having trouble with our tools? Please contact Yanjun Qi and we’ll help you sort it out.
01 Oct 2009
Title: Semi-Supervised Sequence Labeling with Self-Learned Feature
- authors: Yanjun
Qi,
Pavel
P
Kuksa,
Ronan
Collobert, Kunihiko
Sadamasa,
Koray
Kavukcuoglu,
Jason
Weston
Abstract
Typical information extraction (IE) systems can be seen as tasks assigning labels to words in a natural language sequence. The performance is restricted by the availability of labeled words. To tackle this issue, we propose a semi-supervised approach to improve the sequence labeling procedure in IE through a class of algorithms with self-learned features (SLF). A supervised classifier can be trained with annotated text sequences and used to classify each word in a large set of unannotated sentences. By averaging predicted labels over all cases in the unlabeled corpus, SLF training builds class label distribution patterns for each word (or word attribute) in the dictionary and re-trains the current model iteratively adding these distributions as extra word features. Basic SLF models how likely a word could be assigned to target class types. Several extensions are proposed, such as learning words’ class boundary distributions. SLF exhibits robust and scalable behaviour and is easy to tune. We applied this approach on four classical IE tasks: named entity recognition (German and English), part-of-speech tagging (English) and one gene name recognition corpus. Experimental results show effective improvements over the supervised baselines on all tasks. In addition, when compared with the closely related self-training idea, this approach shows favorable advantages.
Citations
@inproceedings{qi2009semi,
title={Semi-supervised sequence labeling with self-learned features},
author={Qi, Yanjun and Kuksa, Pavel and Collobert, Ronan and Sadamasa, Kunihiko and Kavukcuoglu, Koray and Weston, Jason},
booktitle={2009 Ninth IEEE International Conference on Data Mining},
pages={428--437},
year={2009},
organization={IEEE}
}




Having trouble with our tools? Please contact Yanjun Qi and we’ll help you sort it out.
01 Feb 2009
Title: Semi-supervised multi-task learning for predicting interactions between HIV-1 and human proteins
- authors: Yanjun Qi, Oznur Tastan, Jaime G. Carbonell, Judith Klein-Seetharaman, Jason Weston
Abstract
-
Motivation: Protein–protein interactions (PPIs) are critical for virtually every biological function. Recently, researchers suggested to use supervised learning for the task of classifying pairs of proteins as interacting or not. However, its performance is largely restricted by the availability of truly interacting proteins (labeled). Meanwhile, there exists a considerable amount of protein pairs where an association appears between two partners, but not enough experimental evidence to support it as a direct interaction (partially labeled).
-
Results: We propose a semi-supervised multi-task framework for predicting PPIs from not only labeled, but also partially labeled reference sets. The basic idea is to perform multi-task learning on a supervised classification task and a semi-supervised auxiliary task. The supervised classifier trains a multi-layer perceptron network for PPI predictions from labeled examples. The semi-supervised auxiliary task shares network layers of the supervised classifier and trains with partially labeled examples. Semi-supervision could be utilized in multiple ways. We tried three approaches in this article, (i) classification (to distinguish partial positives with negatives); (ii) ranking (to rate partial positive more likely than negatives); (iii) embedding (to make data clusters get similar labels). We applied this framework to improve the identification of interacting pairs between HIV-1 and human proteins. Our method improved upon the state-of-the-art method for this task indicating the benefits of semi-supervised multi-task learning using auxiliary information.
Citations
@article{qi2010semi,
title={Semi-supervised multi-task learning for predicting interactions between HIV-1 and human proteins},
author={Qi, Yanjun and Tastan, Oznur and Carbonell, Jaime G and Klein-Seetharaman, Judith and Weston, Jason},
journal={Bioinformatics},
volume={26},
number={18},
pages={i645--i652},
year={2010},
publisher={Oxford University Press}
}


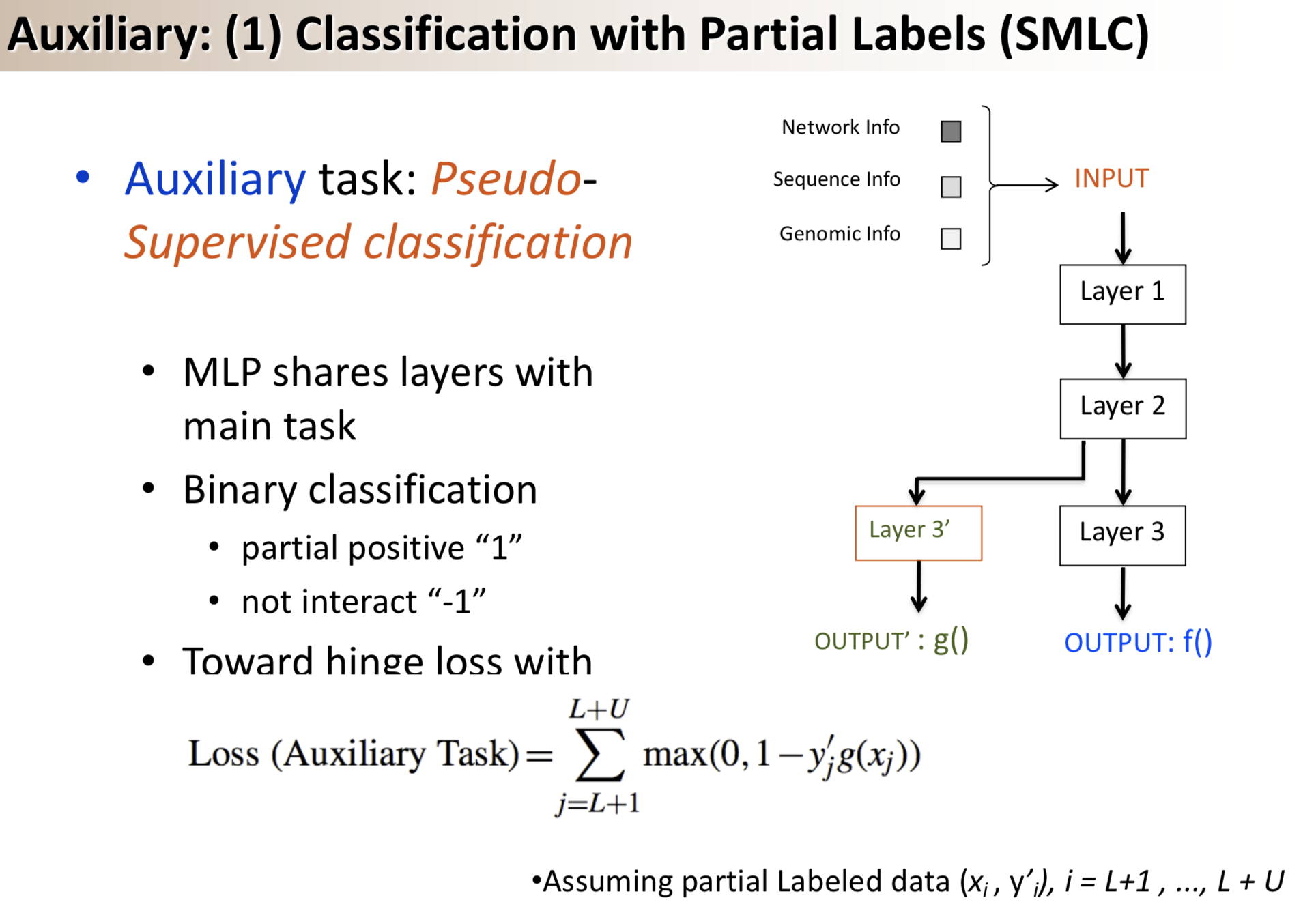
Having trouble with our tools? Please contact Yanjun Qi and we’ll help you sort it out.
