We move the blogs on this research front to our Group-Blog-Site at https://qdata.github.io/qdata-page/ since 2021.
General Background
Biology and medicine are rapidly becoming data-intensive. A recent comparison of genomics with social media, online videos, and other data-intensive disciplines suggests that genomics alone will equal or surpass other fields in data generation and analysis within the next decade. The volume and complexity of these data present new opportunities, but also pose new challenges. Data sets are complex, often ill-understood and grow at a a faster scale than computational capabilities. Problems of this nature may be particularly well-suited to deep learning techniques (see Opportunities and obstacles for deep learning in biology and medicine). The website introduces a suite of deep learning tools we have developed for learning patterns and making predictions on biomedical data (mostly from functional genomics).
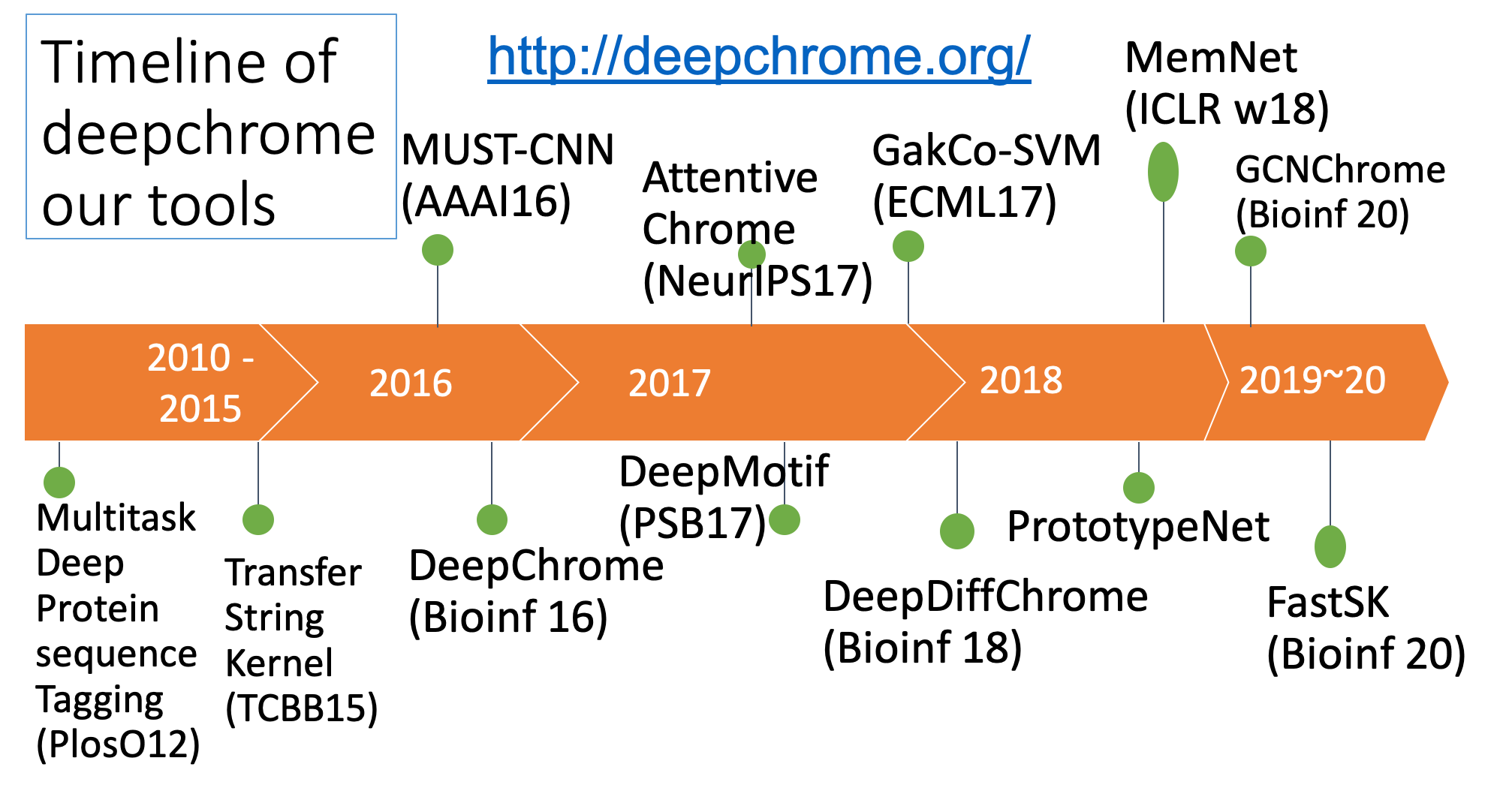
Summary of our tasks and tools
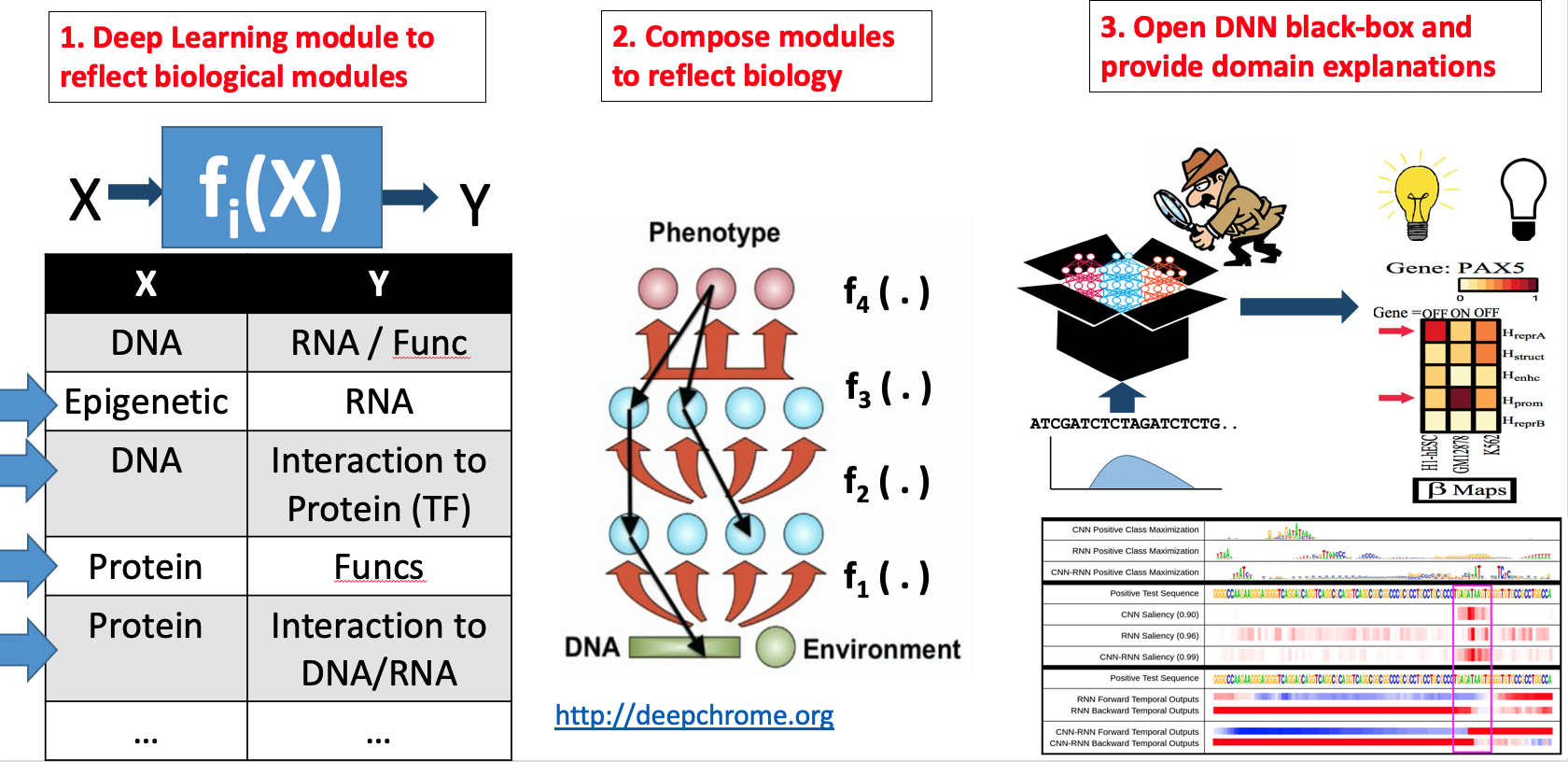
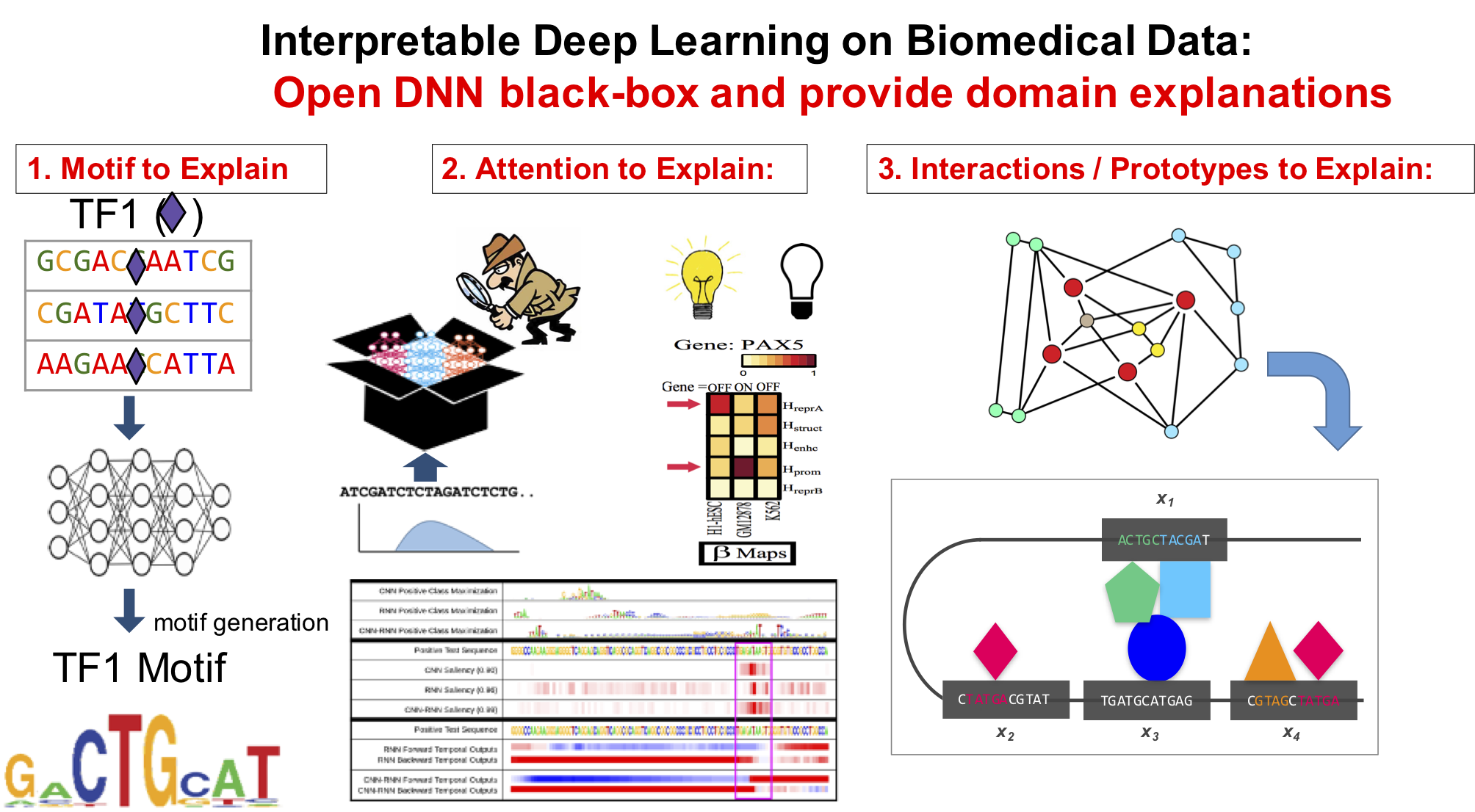
Our technical focus in this direction center on making DNN interpretable.
Background of Learning: Representation Learning and Deep Learning
The performance of machine learning algorithms is largely dependent on the data representation (or features) on which they are applied. Deep learning aims at discovering learning algorithms that can find multiple levels of representations directly from data, with higher levels representing more abstract concepts. In recent years, the field of deep learning has lead to groundbreaking performance in many applications such as computer vision, speech understanding, natural language processing, and computational biology.
Background of Biology Relevant to Our DeepChrome and AttentiveChrome tools
DNA is a long string of paired chemical units that fall into four different types (ATCG). DNA carries information organized into units such as genes. The set of DNA in a cell is called its genome.
Gene regulation is the process of how a cell controls which genes in its genome are turned on (expressed) or off (not-expressed). The human body contains hundreds of different cell types, from liver cells to blood cells to neurons. Although these cells include the same set of DNA information, they function differently.
The regulation of different genes controls the destiny and function of each cell.
In addition to DNA sequence information, many factors, especially those in its environment (i.e., chromatin) can affect which genes the cell expresses. Our tools aim to invent novel machine learning, especially deep learning based architecture to learn from data how different chromatin factors, DNA sequences and other environmental factors influence gene expression in a cell. Such understanding of gene regulation can enable new insights into principles of life, the study of disease, and drug development.
‘Chromatin’ denotes DNA and its organizing proteins. The complex of DNA, histones, and other structural proteins is called chromatin. A cell uses specialized proteins to organize DNA in a condensed structure. These proteins include histones, which form `bead’-like structures that DNA wraps around, in turn organizing and making the DNA more compact. An important aspect of histone proteins is that they are prone to chemical modifications that can change the spatial arrangement of DNA, resulting in certain DNA regions becoming accessible or restricted and therefore affecting expressions of genes in the neighborhood region. Researchers have established the ‘Histone Code Hypothesis’ that explores the role of histone modifications in controlling gene regulation. Unlike genetic mutations, chromatin changes such as histone modifications are potentially reversible. This crucial difference makes the understanding of how chromatin factors determine gene regulation even more impactful because the knowledge can help developing drugs targeting genetic diseases.
At the whole genome level, researchers are trying to chart the locations and intensities of all the chemical modifications, referred to as marks, over the chromatin. In biology this field is called epigenetics. ‘Epi’ in Greek means over. The epigenome in a cell is the set of chemical modifications over the chromatin that alter gene expression.
Recent advances in next-generation sequencing have allowed biologists to profile a significant amount of gene expression and chromatin patterns as signals (or read counts) across many cell types covering the full human genome.
These datasets have been made available through large-scale repositories, the latest being the Roadmap Epigenome Project (REMC, publicly available).
REMC recently released 2,804 genome-wide datasets, among which 166 datasets are gene expression reads (RNA-Seq datasets) and the rest are signal reads of various chromatin marks across 100 different `normal’ human cells/tissues (1,821 datasets for histone modification marks).
The fundamental aim of processing and understanding this repository of ‘big’ data is to understand gene regulation. For each cell type, we want to know which chromatin marks are the most important and how they work together in controlling gene expression. However, previous machine learning studies on this task either failed to model spatial dependencies among mark signals or required additional feature analysis to explain the predictions
Contacts:
Have questions or suggestions? Feel free to ask me on Twitter or email me.
Thanks for reading!