SVM-StringKernel (Index of Posts):
We create this categoy of string kernel tools for calculating fast gapped-kmer-string-kernel calculations on biological sequence inputs..
This includes:
01 Jun 2020
Title: FastSK: Fast Sequence Analysis with Gapped String Kernels

Abstract
Gapped k-mer kernels with Support Vector Machines (gkm-SVMs)
have achieved strong predictive performance on regulatory DNA sequences
on modestly-sized training sets. However, existing gkm-SVM algorithms
suffer from the slow kernel computation time, as they depend
exponentially on the sub-sequence feature-length, number of mismatch
positions, and the task’s alphabet size.
In this work, we introduce a fast and scalable algorithm for
calculating gapped k-mer string kernels. Our method, named FastSK,
uses a simplified kernel formulation that decomposes the kernel
calculation into a set of independent counting operations over the
possible mismatch positions. This simplified decomposition allows us
to devise a fast Monte Carlo approximation that rapidly converges.
FastSK can scale to much greater feature lengths, allows us to
consider more mismatches, and is performant on a variety of sequence
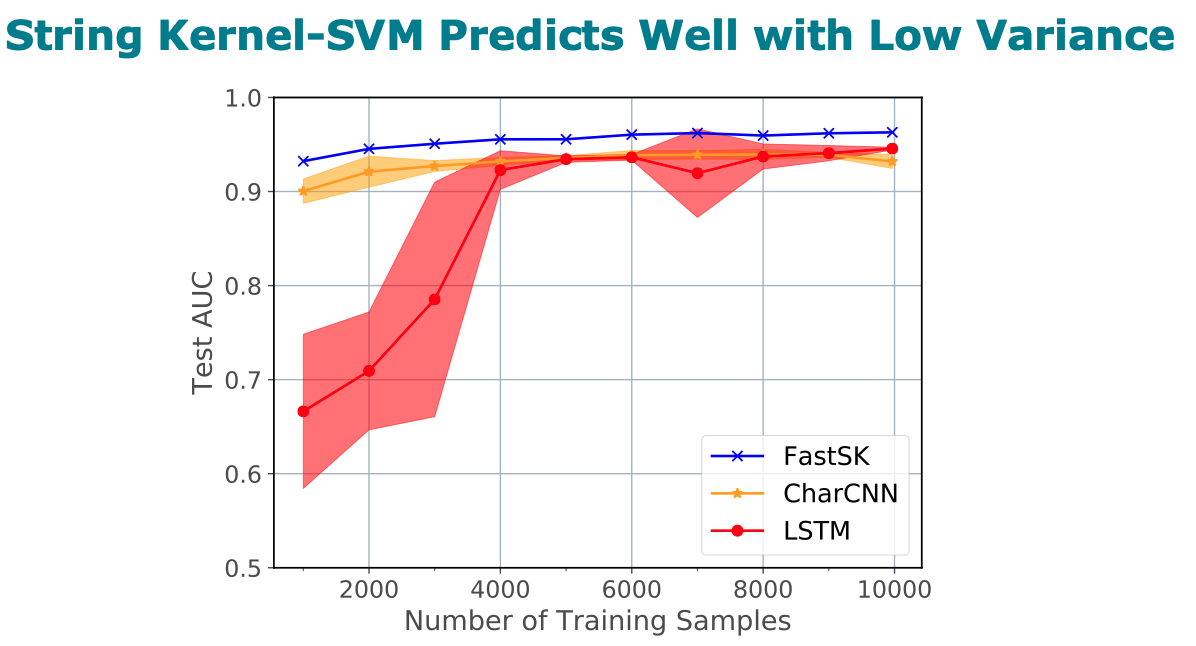
analysis tasks. On 10 DNA transcription factor binding site (TFBS)
prediction datasets, FastSK consistently matches or outperforms the
state-of-the-art gkmSVM-2.0 algorithms in AUC, while achieving
average speedups in kernel computation of 100 times and speedups of
800 times for large feature lengths. We further show that FastSK
outperforms character-level recurrent and convolutional neural
networks across all 10 TFBS tasks. We then extend FastSK to 7
English medical named entity recognition datasets and 10 protein
remote homology detection datasets. FastSK consistently matches or
outperforms these baselines.
Our algorithm is available as a Python package and as C++ source code.
(Available for download at https://github.com/Qdata/FastSK/.
Install with the command make or pip install)
Citations
@article{10.1093/bioinformatics/btaa817,
author = {Blakely, Derrick and Collins, Eamon and Singh, Ritambhara and Norton, Andrew and Lanchantin, Jack and Qi, Yanjun},
title = "{FastSK: fast sequence analysis with gapped string kernels}",
journal = {Bioinformatics},
volume = {36},
number = {Supplement_2},
pages = {i857-i865},
year = {2020},
month = {12},
issn = {1367-4803},
doi = {10.1093/bioinformatics/btaa817},
url = {https://doi.org/10.1093/bioinformatics/btaa817},
eprint = {https://academic.oup.com/bioinformatics/article-pdf/36/Supplement\_2/i857/35337038/btaa817.pdf},
}
Having trouble with our tools? Please contact Yanjun Qi and we’ll help you sort it out.
20 Jun 2016
Abstract:
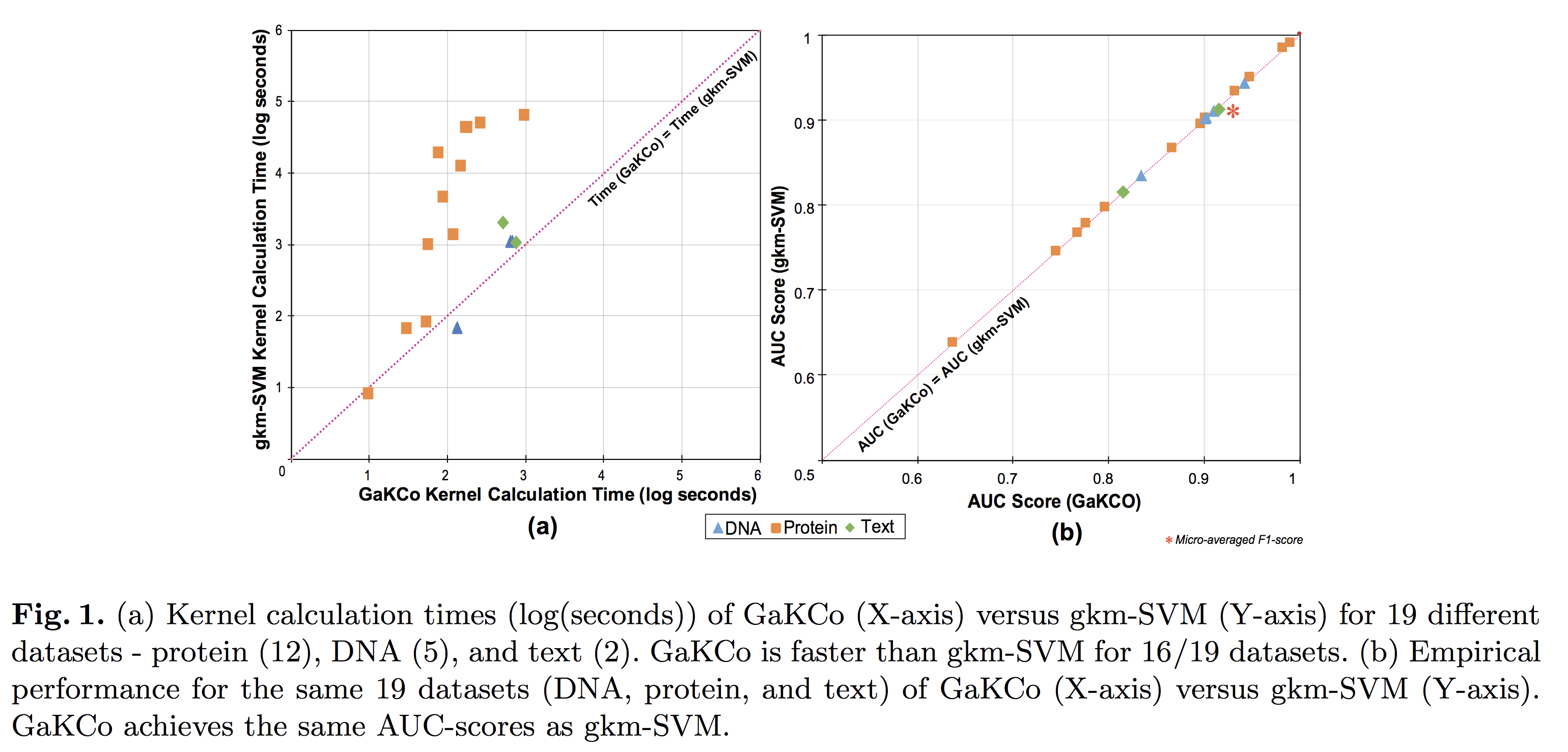
String Kernel (SK) techniques, especially those using gapped k-mers as features (gk), have obtained great success in classifying sequences like DNA, protein, and text. However, the state-of-the-art gk-SK runs extremely slow when we increase the dictionary size (Σ) or allow more mismatches (M). This is because current gk-SK uses a trie-based algorithm to calculate co-occurrence of mismatched substrings resulting in a time cost proportional to O(ΣM). We propose a \textbf{fast} algorithm for calculating \underline{Ga}pped k-mer \underline{K}ernel using \underline{Co}unting (GaKCo). GaKCo uses associative arrays to calculate the co-occurrence of substrings using cumulative counting. This algorithm is fast, scalable to larger Σ and M, and naturally parallelizable. We provide a rigorous asymptotic analysis that compares GaKCo with the state-of-the-art gk-SK. Theoretically, the time cost of GaKCo is independent of the ΣM term that slows down the trie-based approach. Experimentally, we observe that GaKCo achieves the same accuracy as the state-of-the-art and outperforms its speed by factors of 2, 100, and 4, on classifying sequences of DNA (5 datasets), protein (12 datasets), and character-based English text (2 datasets), respectively.
Citations
@inproceedings{singh_gakco:_2017,
location = {Cham},
title = {GaKCo: A Fast Gapped k-mer String Kernel Using Counting},
isbn = {978-3-319-71249-9},
pages = {356--373},
booktitle = {Machine Learning and Knowledge Discovery in Databases},
publisher = {Springer International Publishing},
author = {Singh, Ritambhara and Sekhon, Arshdeep and Kowsari, Kamran and Lanchantin, Jack and Wang, Beilun and Qi, Yanjun},
editor = {Ceci, Michelangelo and Hollmén, Jaakko and Todorovski, Ljupčo and Vens, Celine and Džeroski, Sašo},
date = {2017}
}
Having trouble with our tools? Please contact Rita and we’ll help you sort it out.
15 Jun 2016
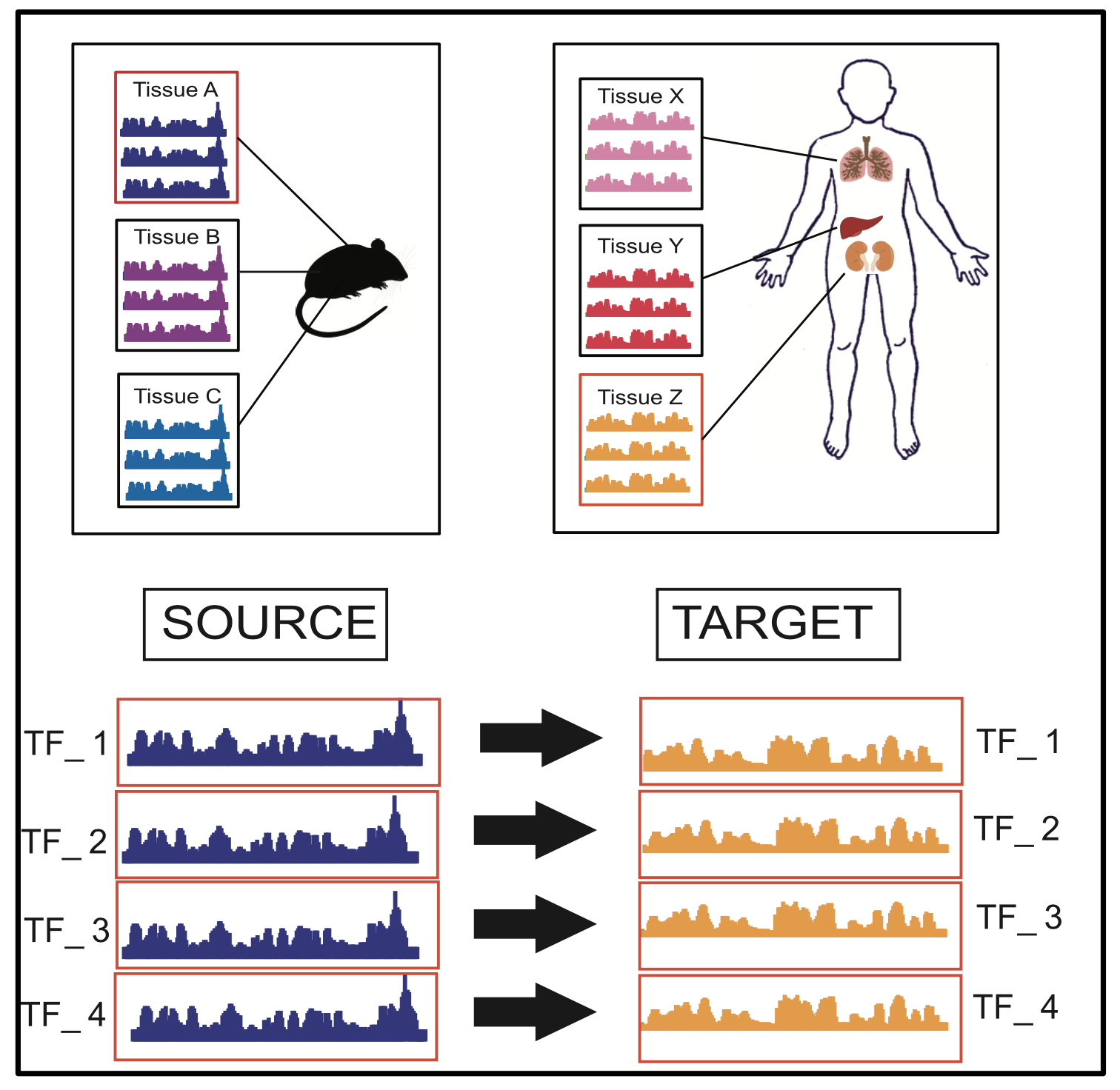
Tool TSK: Transfer String Kernel for Cross-Context DNA-Protein Binding Prediction
Abstract
Through sequence-based classification, this paper tries to accurately predict the DNA binding sites of transcription factors (TFs) in an unannotated cellular context. Related methods in the literature fail to perform such predictions accurately, since they do not consider sample distribution shift of sequence segments from an annotated (source) context to an unannotated (target) context. We, therefore, propose a method called “Transfer String Kernel” (TSK) that achieves improved prediction of transcription factor binding site (TFBS) using knowledge transfer via cross-context sample adaptation. TSK maps sequence segments to a high-dimensional feature space using a discriminative mismatch string kernel framework. In this high-dimensional space, labeled examples of the source context are re-weighted so that the revised sample distribution matches the target context more closely. We have experimentally verified TSK for TFBS identifications on fourteen different TFs under a cross-organism setting. We find that TSK consistently outperforms the state-of the-art TFBS tools, especially when working with TFs whose binding sequences are not conserved across contexts. We also demonstrate the generalizability of TSK by showing its cutting-edge performance on a different set of cross-context tasks for the MHC peptide binding predictions.
Citations
@article{singh2016transfer,
title={Transfer String Kernel for Cross-Context DNA-Protein Binding Prediction},
author={Singh, Ritambhara and Lanchantin, Jack and Robins, Gabriel and Qi, Yanjun},
journal={IEEE/ACM Transactions on Computational Biology and Bioinformatics},
year={2016},
publisher={IEEE}
}
Having trouble with our tools? Please contact Rita and we’ll help you sort it out.
01 Mar 2010
Title: Systems and methods for semi-supervised relationship extraction
- authors: Qi, Yanjun and Bai, Bing and Ning, Xia and Kuksa, Pavel
- PDF
-
Talk: Slide
- Abstract
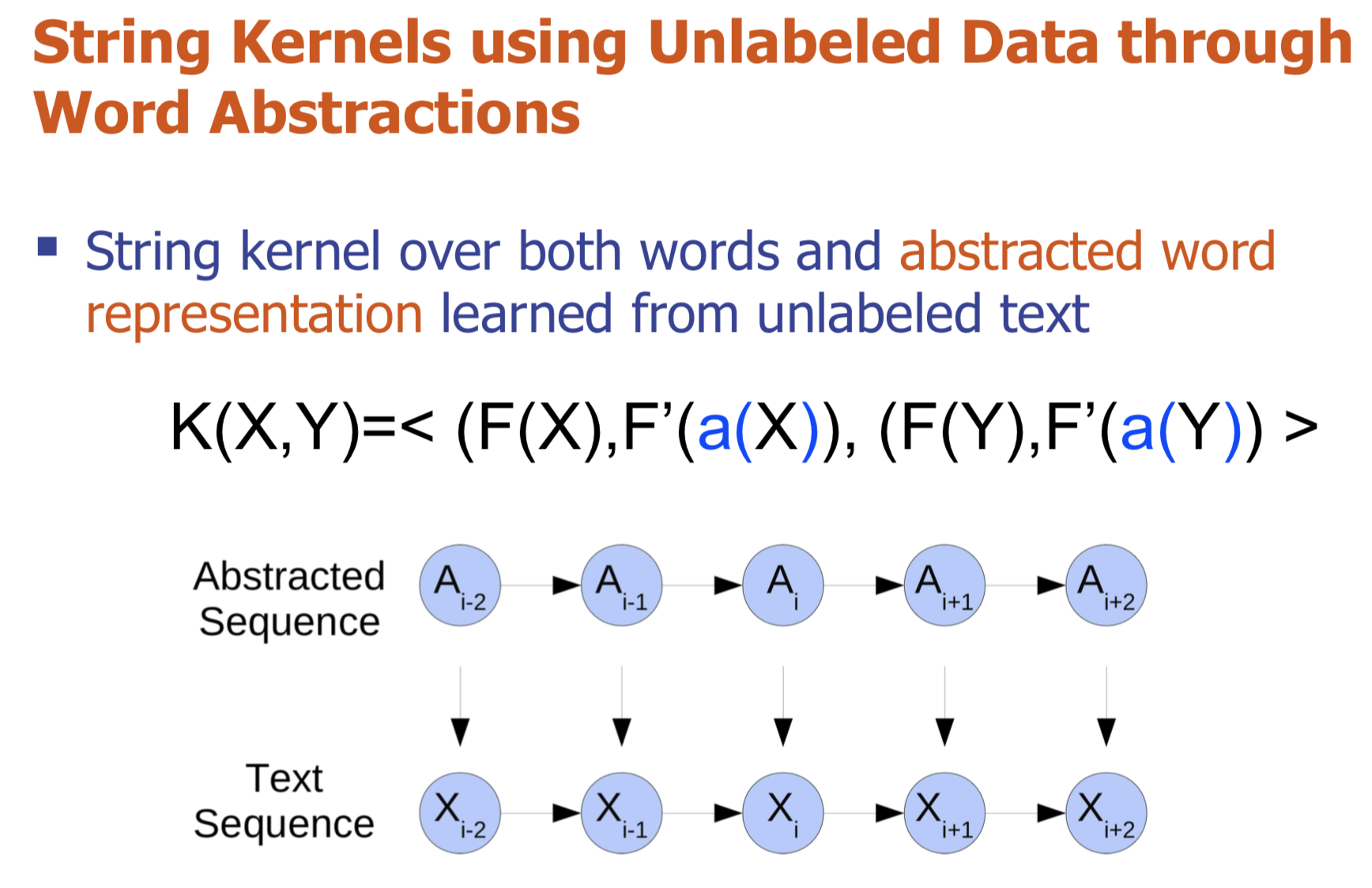
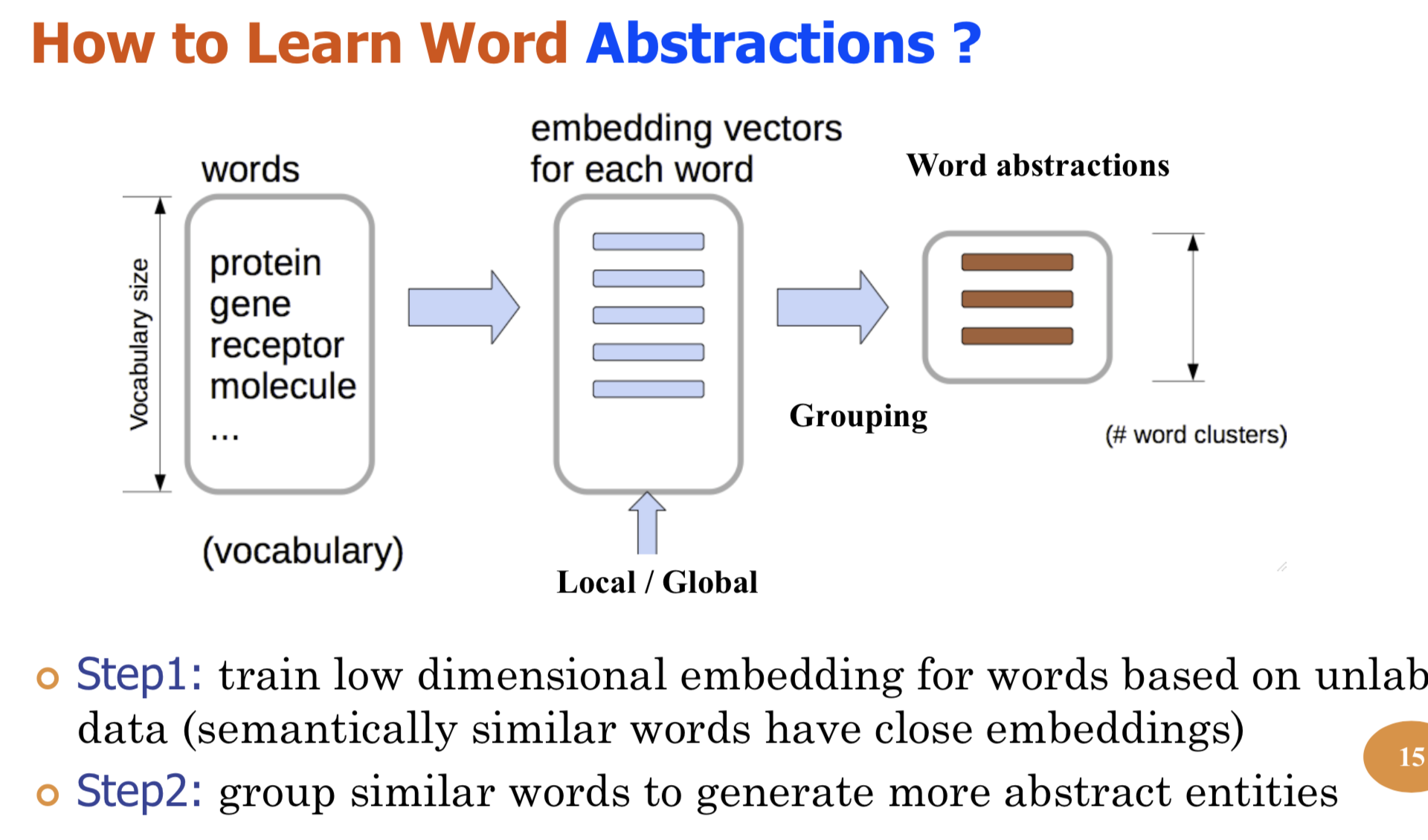
Bio-relation extraction (bRE), an important goal in bio-text mining, involves subtasks identifying relationships between bio-entities in text at multiple levels, e.g., at the article, sentence or relation level. A key limitation of current bRE systems is that they are restricted by the availability of annotated corpora. In this work we introduce a semi-supervised approach that can tackle multi-level bRE via string comparisons with mismatches in the string kernel framework. Our string kernel implements an abstraction step, which groups similar words to generate more abstract entities, which can be learnt with unlabeled data. Specifically, two unsupervised models are proposed to capture contextual (local or global) semantic similarities between words from a large unannotated corpus. This Abstraction-augmented String Kernel (ASK) allows for better generalization of patterns learned from annotated data and provides a unified framework for solving bRE with multiple degrees
of detail. ASK shows effective improvements over classic string kernels
on four datasets and achieves state-of-the-art bRE performance without
the need for complex linguistic features.
-
PDF
- Talk: Slide
-
URL More
- Abstract
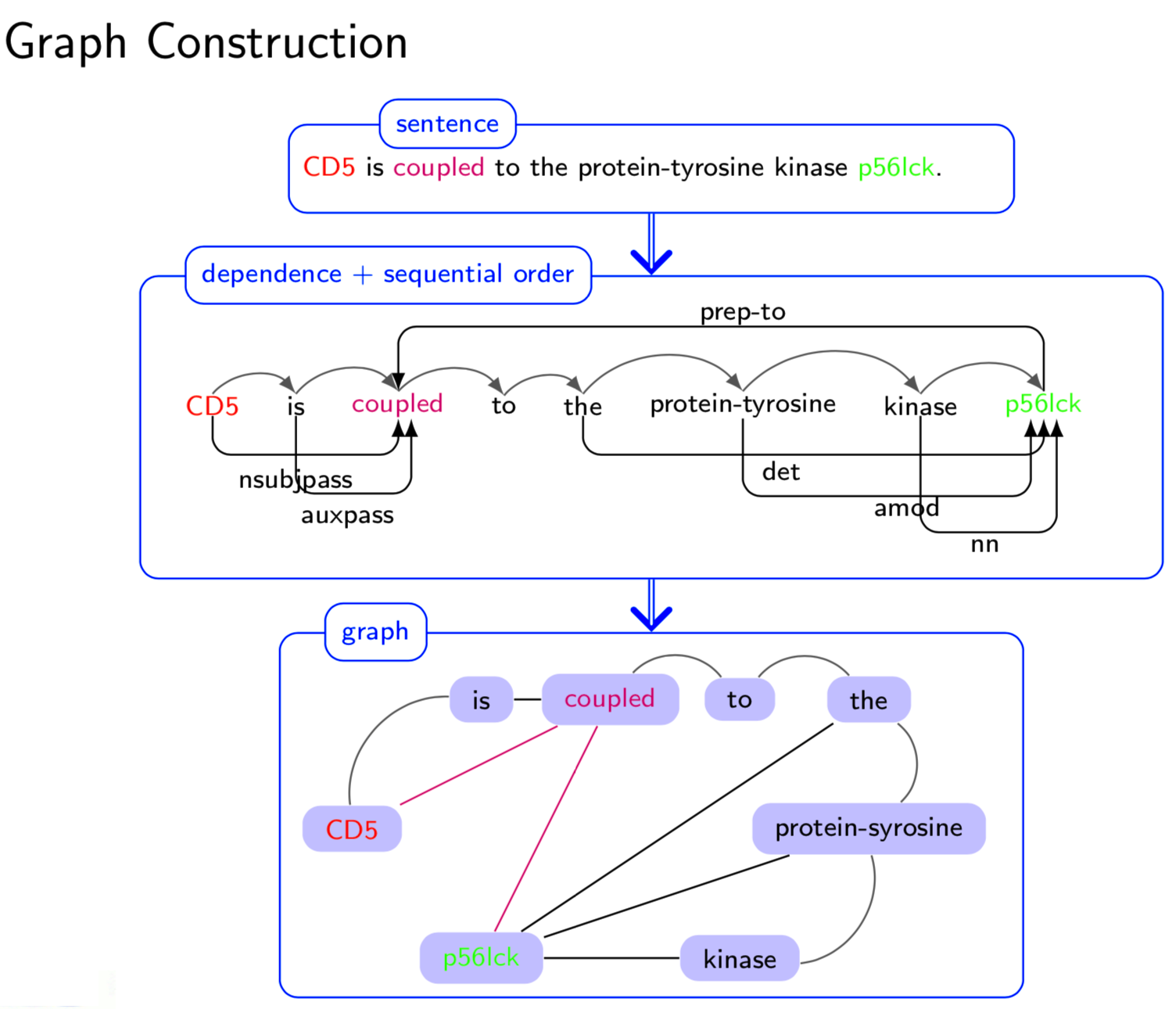
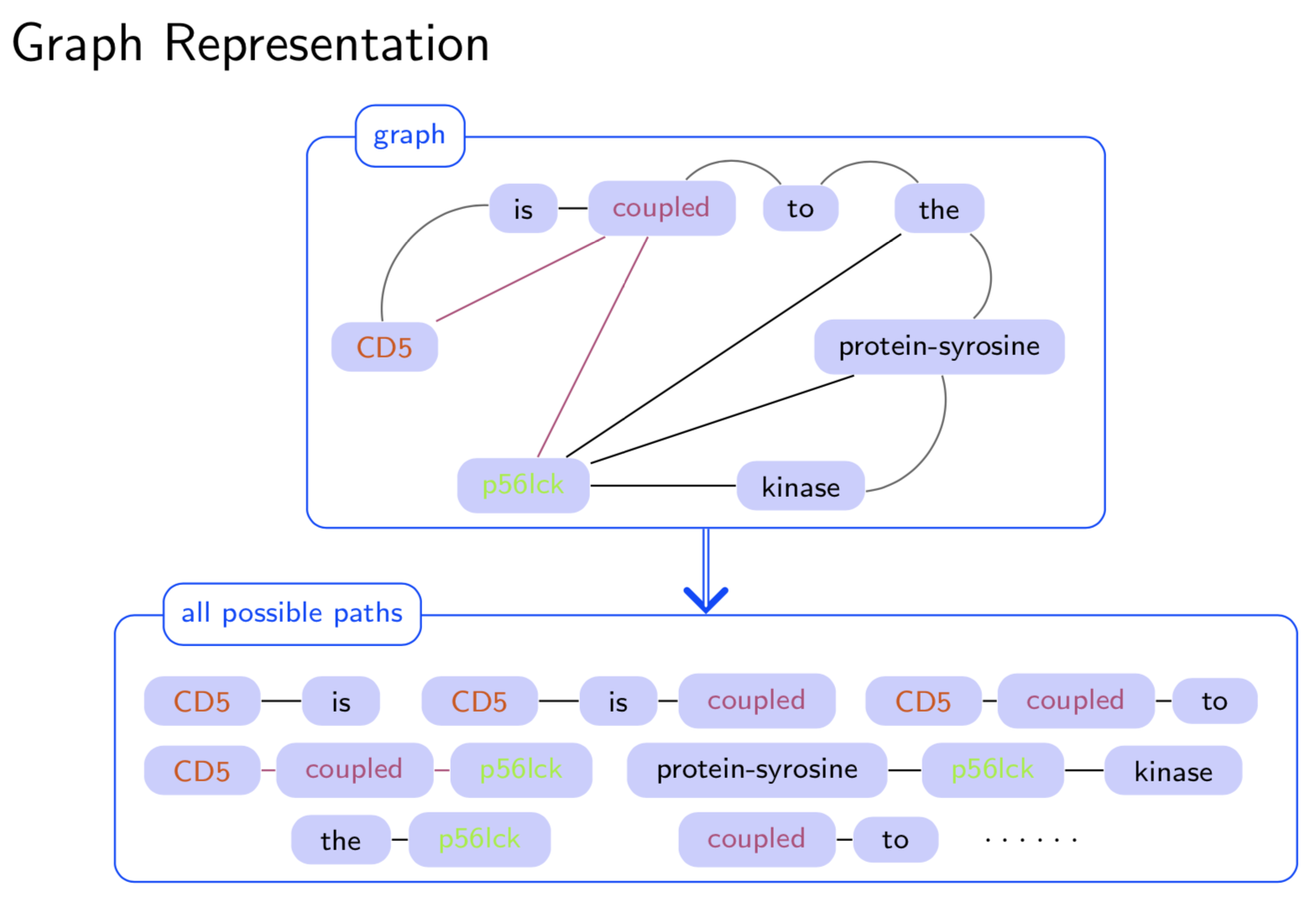
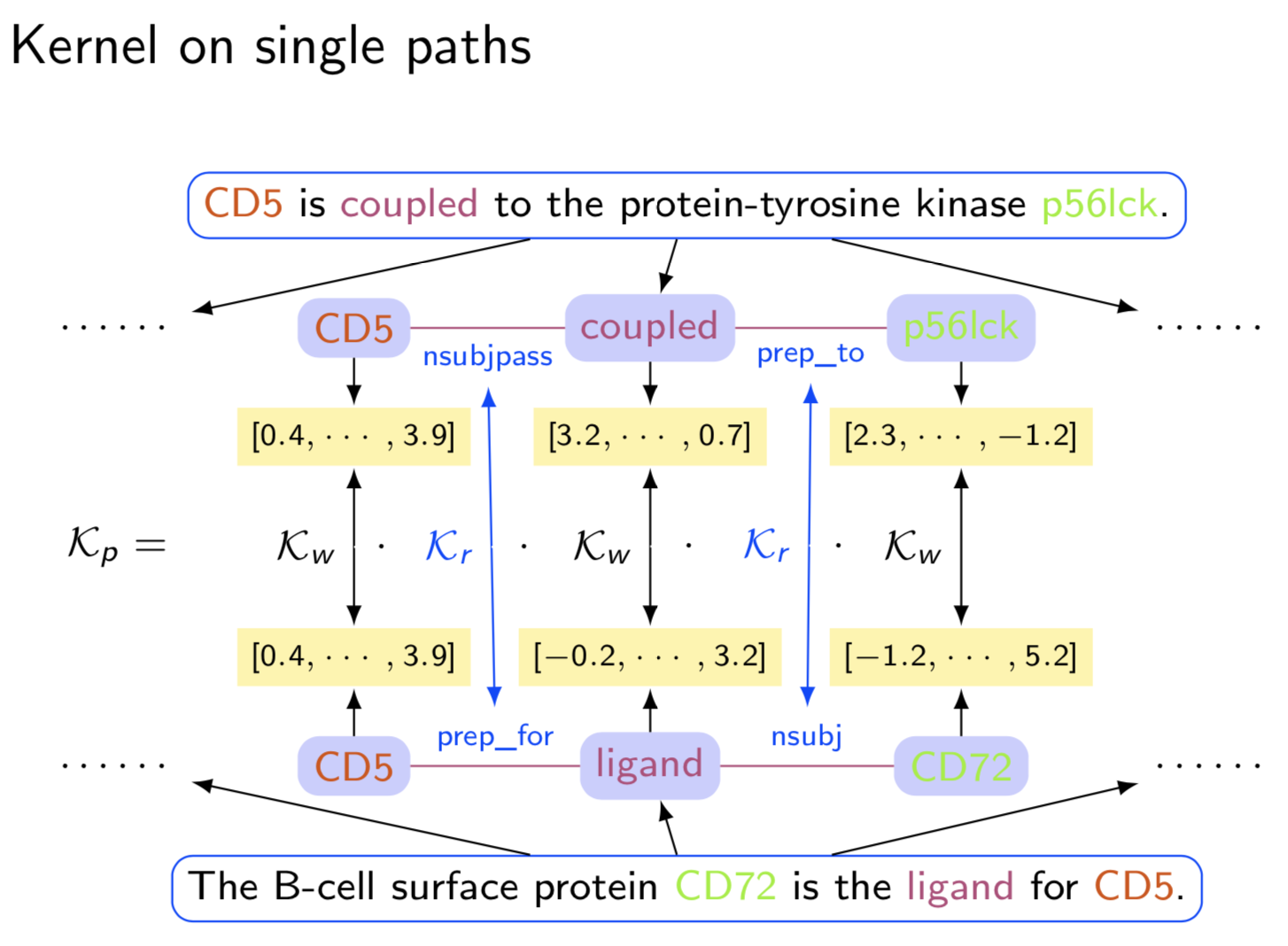
Extracting semantic relations between entities is an important step towards automatic text understanding. In this paper, we propose a novel Semi-supervised Convolution Graph Kernel (SCGK) method for semantic Relation Extraction (RE) from natural language. By encoding English sentences as dependence graphs among words, SCGK computes kernels (similarities) between sentences using a convolution strategy, i.e., calculating similarities over all possible short single paths from two dependence graphs. Furthermore, SCGK adds three semi-supervised strategies in the kernel calculation to incorporate soft-matches between (1) words, (2) grammatical dependencies, and (3) entire sentences, respectively. From a large unannotated corpus, these semi-supervision steps learn to capture contextual semantic patterns of elements in natural sentences, which therefore alleviate the lack of annotated examples in most RE corpora. Through convolutions and multi-level semi-supervisions, SCGK provides a powerful model to encode both syntactic and semantic evidence existing in natural English sentences, which effectively recovers the target relational patterns of interest. We perform extensive experiments on five RE benchmark datasets which aim to identify interaction relations from biomedical literature. Our results demonstrate that SCGK achieves the state-of-the-art performance on the task of semantic relation extraction.
Paper3: Semi-Supervised Bio-Named Entity Recognition with Word-Codebook Learning
- Pavel P. Kuksa, Yanjun Qi,
-
PDF
- Abstract
We describe a novel semi-supervised method called WordCodebook Learning (WCL), and apply it to the task of bionamed entity recognition (bioNER). Typical bioNER systems can be seen as tasks of assigning labels to words in bioliterature text. To improve supervised tagging, WCL learns
a class of word-level feature embeddings to capture word
semantic meanings or word label patterns from a large unlabeled corpus. Words are then clustered according to their
embedding vectors through a vector quantization step, where
each word is assigned into one of the codewords in a codebook. Finally codewords are treated as new word attributes
and are added for entity labeling. Two types of wordcodebook learning are proposed: (1) General WCL, where
an unsupervised method uses contextual semantic similarity of words to learn accurate word representations; (2)
Task-oriented WCL, where for every word a semi-supervised
method learns target-class label patterns from unlabeled
data using supervised signals from trained bioNER model.
Without the need for complex linguistic features, we demonstrate utility of WCL on the BioCreativeII gene name recognition competition data, where WCL yields state-of-the-art
performance and shows great improvements over supervised
baselines and semi-supervised counter peers.
Citations
@INPROCEEDINGS{ecml2010ask,
author = {Pavel P. Kuksa and Yanjun Qi and Bing Bai and Ronan Collobert and
Jason Weston and Vladimir Pavlovic and Xia Ning},
title = {Semi-Supervised Abstraction-Augmented String Kernel for Multi-Level
Bio-Relation Extraction},
booktitle = {ECML},
year = {2010},
note = {Acceptance rate: 106/658 (16%)},
bib2html_pubtype = {Refereed Conference},
}


Having trouble with our tools? Please contact Yanjun Qi and we’ll help you sort it out.