Protein (Index of Posts):
Our deep learning tools that learn to analyze protein sequence inputs..
This includes:
11 Nov 2020
- authors: Jack Lanchantin, Arshdeep Sekhon, Clint Miller, Yanjun Qi
Abstract
The novel coronavirus SARS-CoV-2, which causes Coronavirus disease 2019 (COVID-19), is a significant threat to worldwide public health. Viruses such as SARS-CoV-2 infect the human body by forming interactions between virus proteins and human proteins that compromise normal human protein-protein interactions (PPI). Current in vivo methods to identify PPIs between a novel virus and humans are slow, costly, and difficult to cover the vast interaction space. We propose a novel deep learning architecture designed for in silico PPI prediction and a transfer learning approach to predict interactions between novel virus proteins and human proteins. We show that our approach outperforms the state-of-the-art methods significantly in predicting Virus–Human protein interactions for SARS-CoV-2, H1N1, and Ebola.
Citations
@article {Lanchantin2020.12.14.422772,
author = {Lanchantin, Jack and Sekhon, Arshdeep and Miller, Clint and Qi, Yanjun},
title = {Transfer Learning with MotifTransformers for Predicting Protein-Protein Interactions Between a Novel Virus and Humans},
elocation-id = {2020.12.14.422772},
year = {2020},
doi = {10.1101/2020.12.14.422772},
publisher = {Cold Spring Harbor Laboratory},
URL = {https://www.biorxiv.org/content/early/2020/12/15/2020.12.14.422772},
eprint = {https://www.biorxiv.org/content/early/2020/12/15/2020.12.14.422772.full.pdf},
journal = {bioRxiv}
}
Having trouble with our tools? Please contact Jack and we’ll help you sort it out.
01 Jun 2020
Title: FastSK: Fast Sequence Analysis with Gapped String Kernels
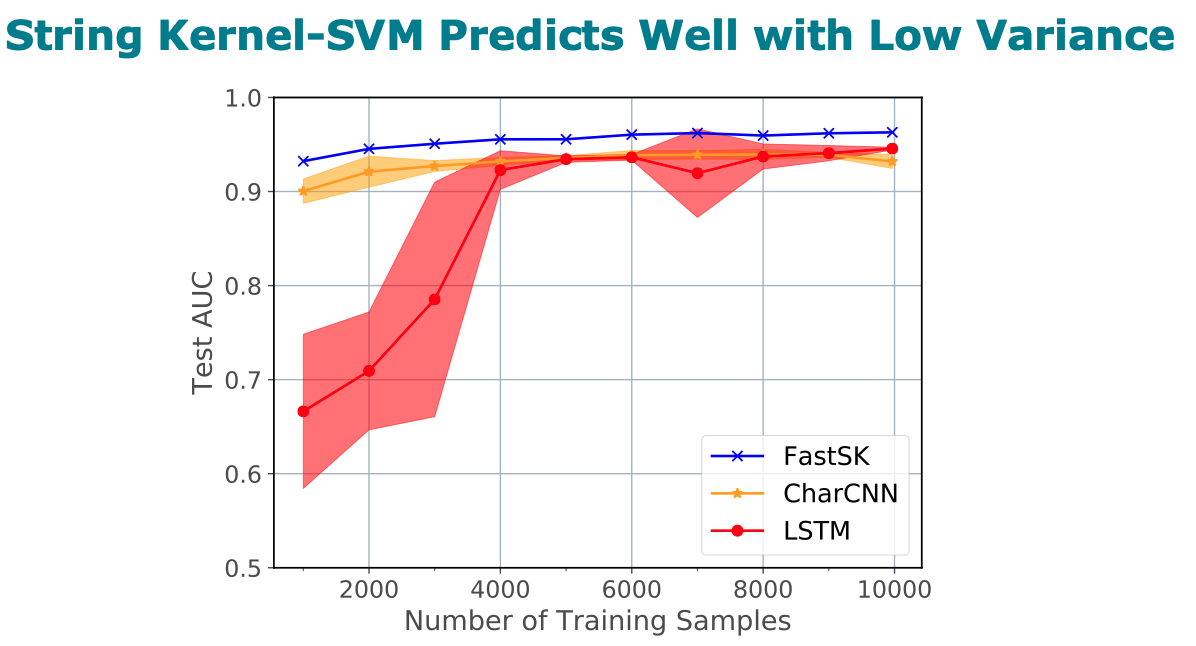

Abstract
Gapped k-mer kernels with Support Vector Machines (gkm-SVMs)
have achieved strong predictive performance on regulatory DNA sequences
on modestly-sized training sets. However, existing gkm-SVM algorithms
suffer from the slow kernel computation time, as they depend
exponentially on the sub-sequence feature-length, number of mismatch
positions, and the task’s alphabet size.
In this work, we introduce a fast and scalable algorithm for
calculating gapped k-mer string kernels. Our method, named FastSK,
uses a simplified kernel formulation that decomposes the kernel
calculation into a set of independent counting operations over the
possible mismatch positions. This simplified decomposition allows us
to devise a fast Monte Carlo approximation that rapidly converges.
FastSK can scale to much greater feature lengths, allows us to
consider more mismatches, and is performant on a variety of sequence
analysis tasks. On 10 DNA transcription factor binding site (TFBS)
prediction datasets, FastSK consistently matches or outperforms the
state-of-the-art gkmSVM-2.0 algorithms in AUC, while achieving
average speedups in kernel computation of 100 times and speedups of
800 times for large feature lengths. We further show that FastSK
outperforms character-level recurrent and convolutional neural
networks across all 10 TFBS tasks. We then extend FastSK to 7
English medical named entity recognition datasets and 10 protein
remote homology detection datasets. FastSK consistently matches or
outperforms these baselines.
Our algorithm is available as a Python package and as C++ source code.
(Available for download at https://github.com/Qdata/FastSK/.
Install with the command make or pip install)
Citations
@article{10.1093/bioinformatics/btaa817,
author = {Blakely, Derrick and Collins, Eamon and Singh, Ritambhara and Norton, Andrew and Lanchantin, Jack and Qi, Yanjun},
title = "{FastSK: fast sequence analysis with gapped string kernels}",
journal = {Bioinformatics},
volume = {36},
number = {Supplement_2},
pages = {i857-i865},
year = {2020},
month = {12},
issn = {1367-4803},
doi = {10.1093/bioinformatics/btaa817},
url = {https://doi.org/10.1093/bioinformatics/btaa817},
eprint = {https://academic.oup.com/bioinformatics/article-pdf/36/Supplement\_2/i857/35337038/btaa817.pdf},
}
Having trouble with our tools? Please contact Yanjun Qi and we’ll help you sort it out.
11 Jun 2015
Abstract
Predicting protein properties such as solvent accessibility and secondary structure from its primary amino acid sequence is an important task in bioinformatics. Recently, a few deep learning models have surpassed the traditional window based multilayer perceptron. Taking inspiration from the image classification domain we propose a deep convolutional neural network architecture, MUST-CNN, to predict protein properties. This architecture uses a novel multilayer shift-and-stitch (MUST) technique to generate fully dense per-position predictions on protein sequences. Our model is significantly simpler than the state-of-the-art, yet achieves better results. By combining MUST and the efficient convolution operation, we can consider far more parameters while retaining very fast prediction speeds. We beat the state-of-the-art performance on two large protein property prediction datasets.
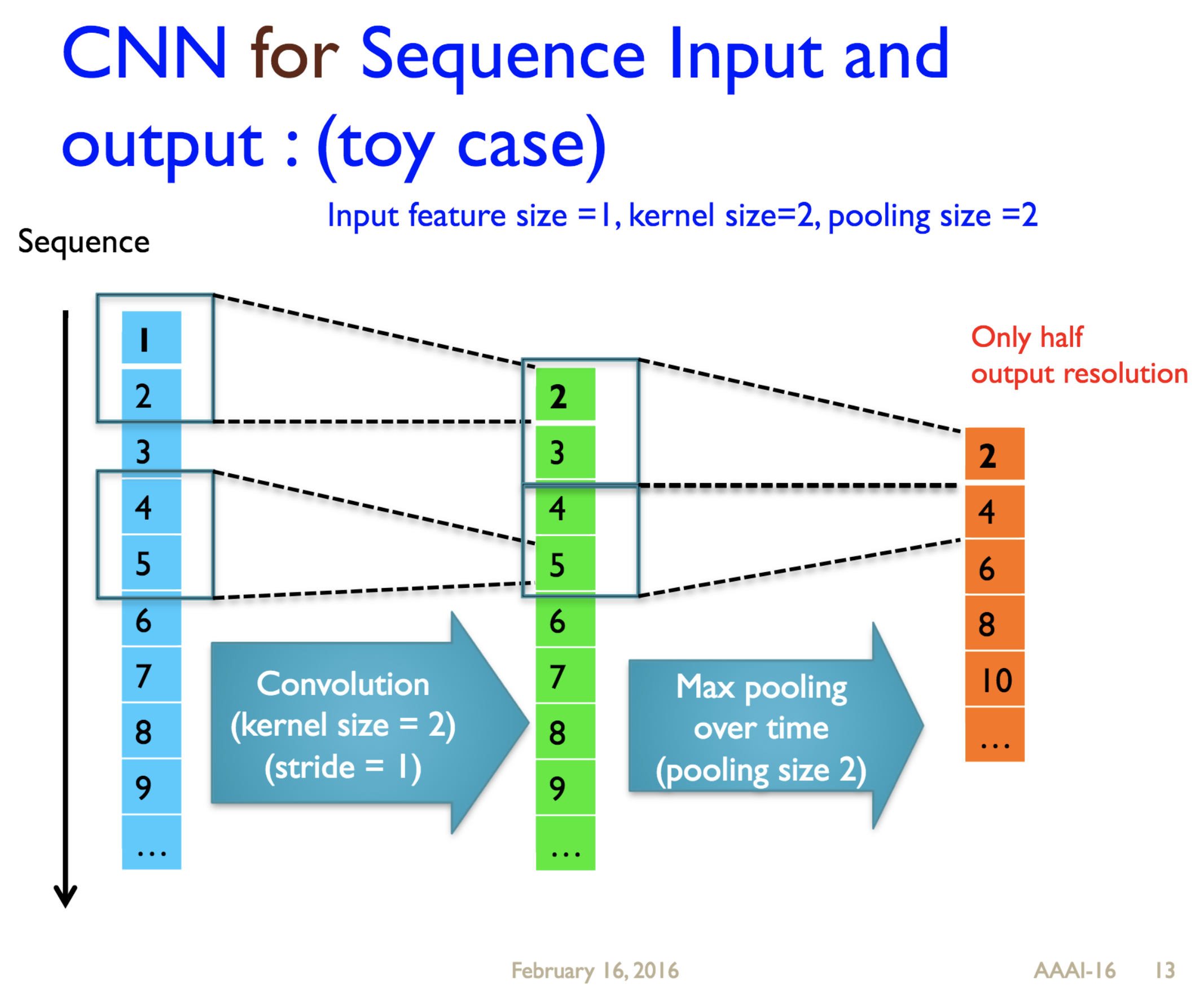
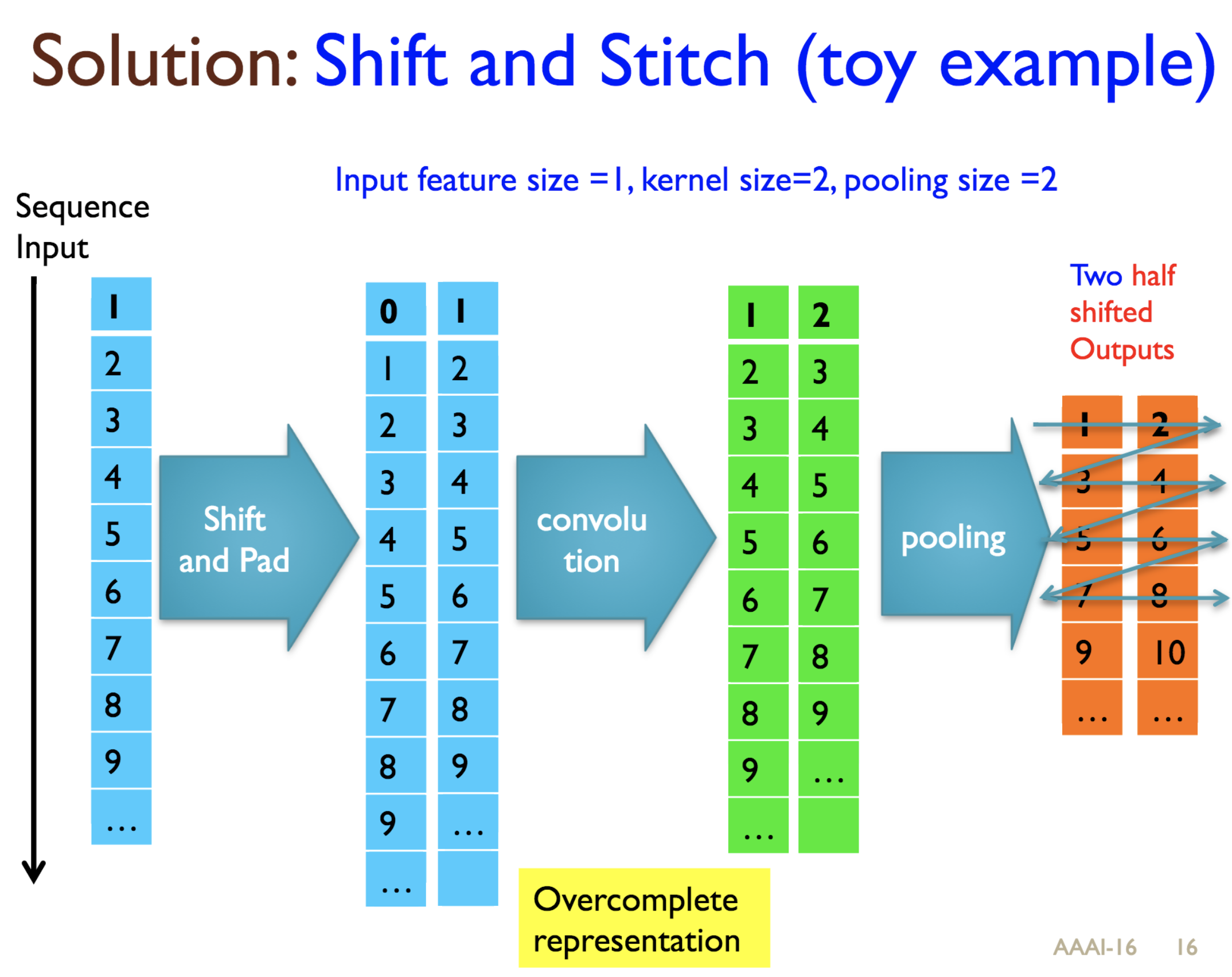
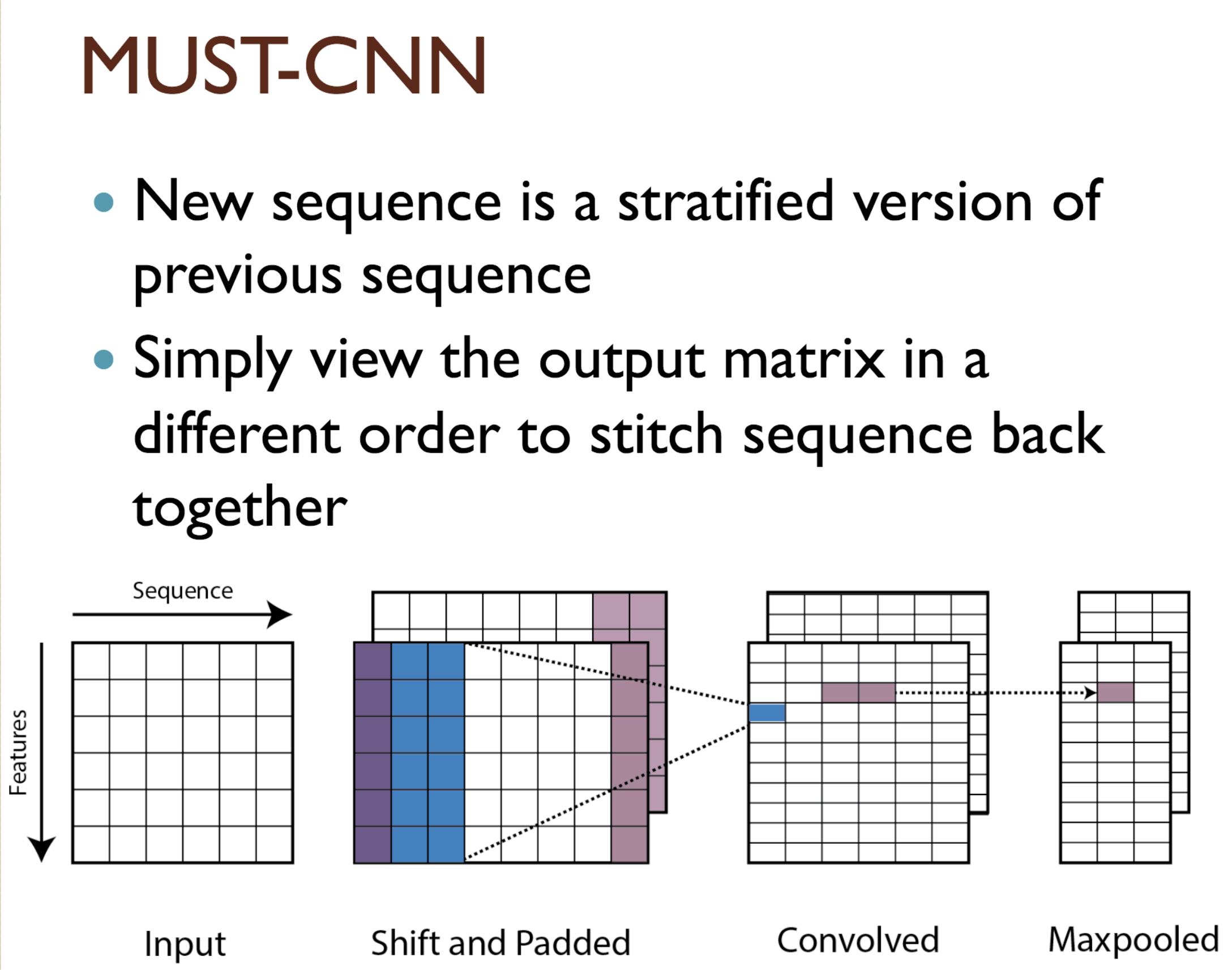

Citations
@inproceedings{lin2016must,
title={MUST-CNN: a multilayer shift-and-stitch deep convolutional architecture for sequence-based protein structure prediction},
author={Lin, Zeming and Lanchantin, Jack and Qi, Yanjun},
booktitle={Proceedings of the Thirtieth AAAI Conference on Artificial Intelligence},
pages={27--34},
year={2016},
organization={AAAI Press}
}
Having trouble with our tools? Please contact Jack and we’ll help you sort it out.
12 Jan 2015
Abstract
A variety of functionally important protein properties, such as secondary structure, transmembrane topology and solvent accessibility, can be encoded as a labeling of amino acids. Indeed, the prediction of such properties from the primary amino acid sequence is one of the core projects of computational biology. Accordingly, a panoply of approaches have been developed for predicting such properties; however, most such approaches focus on solving a single task at a time. Motivated by recent, successful work in natural language processing, we propose to use multitask learning to train a single, joint model that exploits the dependencies among these various labeling tasks. We describe a deep neural network architecture that, given a protein sequence, outputs a host of predicted local properties, including secondary structure, solvent accessibility, transmembrane topology, signal peptides and DNA-binding residues. The network is trained jointly on all these tasks in a supervised fashion, augmented with a novel form of semi-supervised learning in which the model is trained to distinguish between local patterns from natural and synthetic protein sequences. The task-independent architecture of the network obviates the need for task-specific feature engineering. We demonstrate that, for all of the tasks that we considered, our approach leads to statistically significant improvements in performance, relative to a single task neural network approach, and that the resulting model achieves state-of-the-art performance.
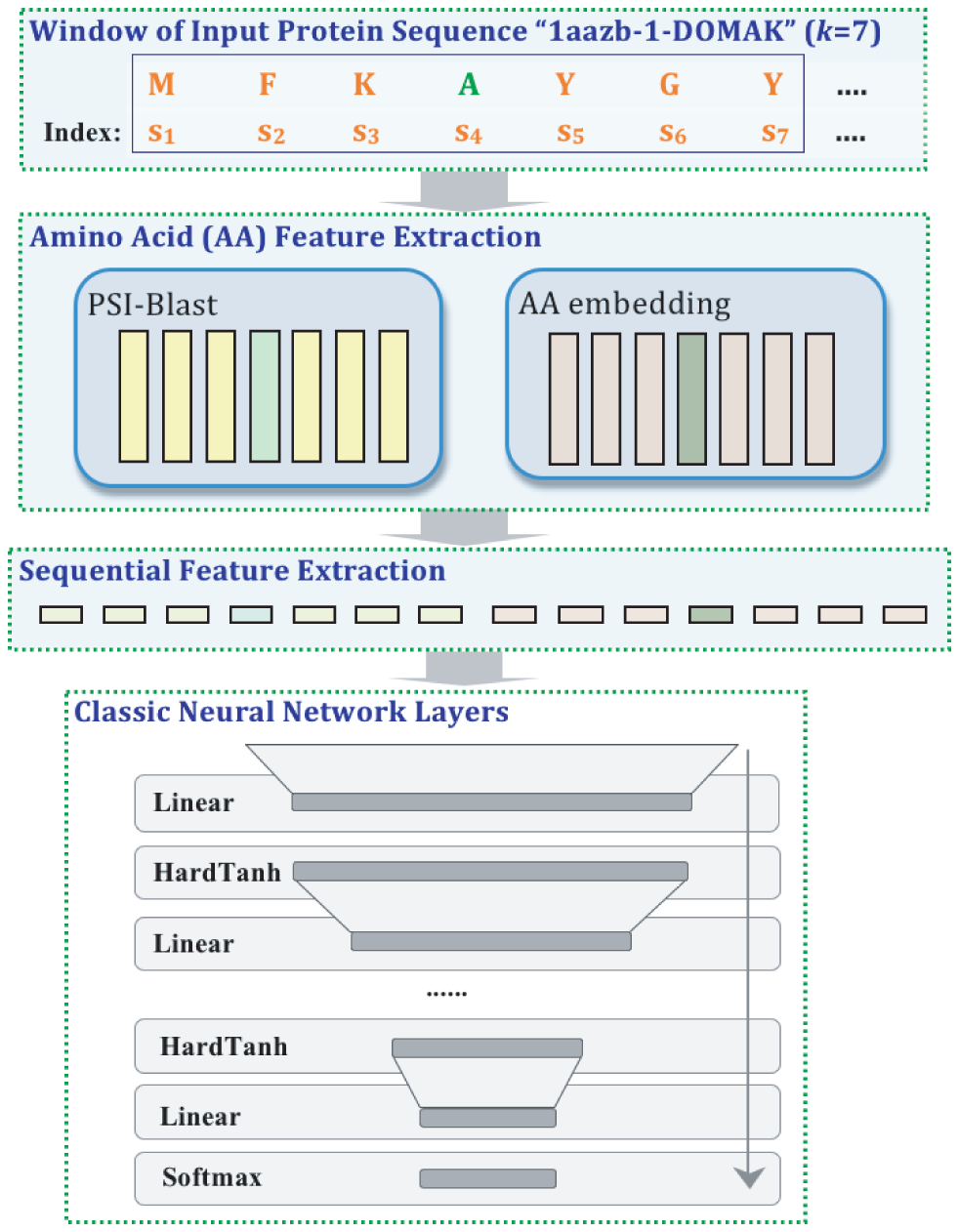
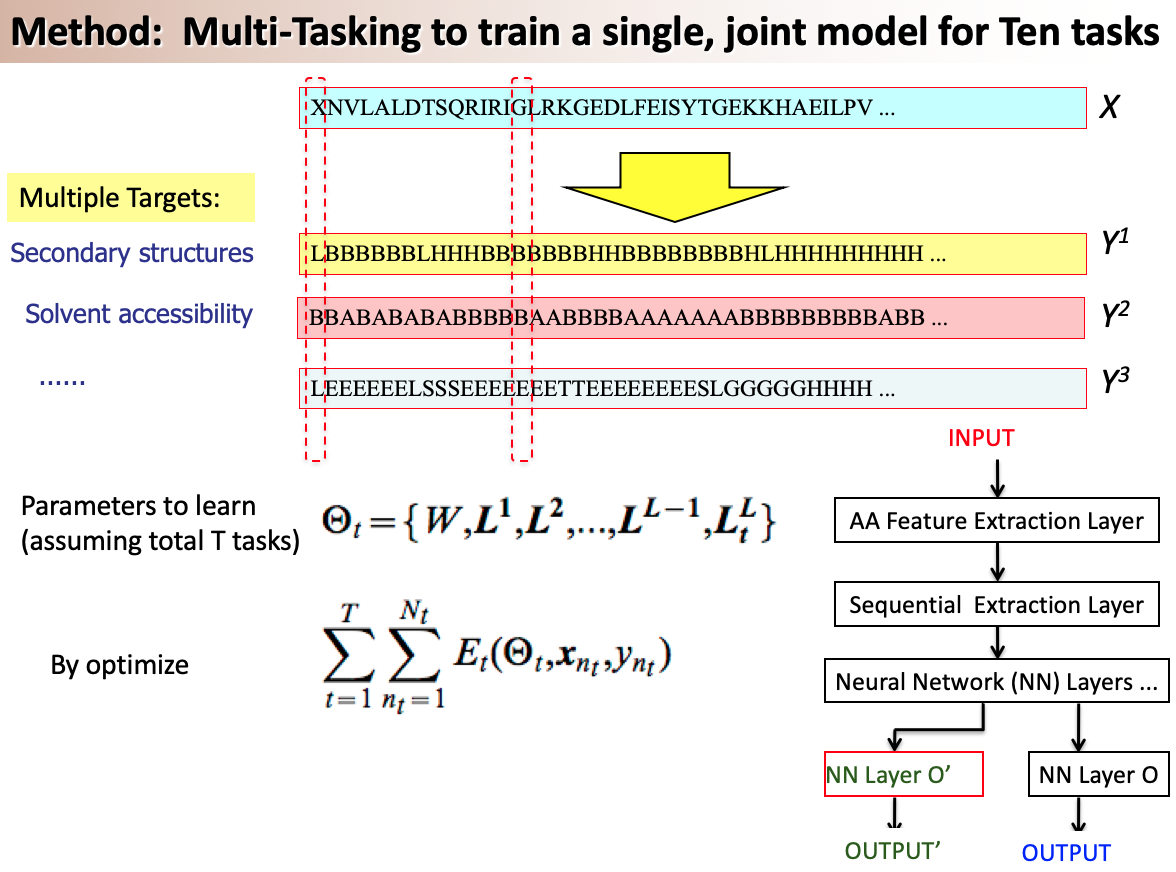
Citations
@article{qi12plosone,
author = {Qi, , Yanjun AND Oja, , Merja AND Weston, , Jason AND Noble, , William Stafford},
journal = {PLoS ONE},
publisher = {Public Library of Science},
title = {A Unified Multitask Architecture for Predicting Local Protein Properties},
year = {2012},
month = {03},
volume = {7},
url = {http://dx.doi.org/10.1371%2Fjournal.pone.0032235},
pages = {e32235},
number = {3},
doi = {10.1371/journal.pone.0032235}
}
Having trouble with our tools? Please contact Jack and we’ll help you sort it out.
01 Oct 2013
- authors: Yanjun Qi, Sujatha Das, Ronan Collobert, Jason Weston
Supplementary Here
Abstract
In this paper we introduce a deep neural network architecture to perform information extraction on character-based sequences,
e.g. named-entity recognition on Chinese text or secondary-structure detection on protein sequences. With a task-independent architecture, the
deep network relies only on simple character-based features, which obviates the need for task-specific feature engineering. The proposed discriminative framework includes three important strategies, (1) a deep
learning module mapping characters to vector representations is included
to capture the semantic relationship between characters; (2) abundant
online sequences (unlabeled) are utilized to improve the vector representation through semi-supervised learning; and (3) the constraints of
spatial dependency among output labels are modeled explicitly in the
deep architecture. The experiments on four benchmark datasets have
demonstrated that, the proposed architecture consistently leads to the
state-of-the-art performance.
Citations
@inproceedings{qi2014deep,
title={Deep learning for character-based information extraction},
author={Qi, Yanjun and Das, Sujatha G and Collobert, Ronan and Weston, Jason},
booktitle={European Conference on Information Retrieval},
pages={668--674},
year={2014},
organization={Springer}
}
Having trouble with our tools? Please contact Yanjun Qi and we’ll help you sort it out.
12 Jan 2013
Abstract
A variety of functionally important protein properties, such as secondary structure, transmembrane topology and solvent accessibility, can be encoded as a labeling of amino acids. Indeed, the prediction of such properties from the primary amino acid sequence is one of the core projects of computational biology. Accordingly, a panoply of approaches have been developed for predicting such properties; however, most such approaches focus on solving a single task at a time. Motivated by recent, successful work in natural language processing, we propose to use multitask learning to train a single, joint model that exploits the dependencies among these various labeling tasks. We describe a deep neural network architecture that, given a protein sequence, outputs a host of predicted local properties, including secondary structure, solvent accessibility, transmembrane topology, signal peptides and DNA-binding residues. The network is trained jointly on all these tasks in a supervised fashion, augmented with a novel form of semi-supervised learning in which the model is trained to distinguish between local patterns from natural and synthetic protein sequences. The task-independent architecture of the network obviates the need for task-specific feature engineering. We demonstrate that, for all of the tasks that we considered, our approach leads to statistically significant improvements in performance, relative to a single task neural network approach, and that the resulting model achieves state-of-the-art performance.
Citations
@article{qi12plosone,
author = {Qi, , Yanjun AND Oja, , Merja AND Weston, , Jason AND Noble, , William Stafford},
journal = {PLoS ONE},
publisher = {Public Library of Science},
title = {A Unified Multitask Architecture for Predicting Local Protein Properties},
year = {2012},
month = {03},
volume = {7},
url = {http://dx.doi.org/10.1371%2Fjournal.pone.0032235},
pages = {e32235},
number = {3},
doi = {10.1371/journal.pone.0032235}
}
Having trouble with our tools? Please contact Jack and we’ll help you sort it out.
01 Feb 2009
Title: Semi-supervised multi-task learning for predicting interactions between HIV-1 and human proteins
- authors: Yanjun Qi, Oznur Tastan, Jaime G. Carbonell, Judith Klein-Seetharaman, Jason Weston
Abstract
-
Motivation: Protein–protein interactions (PPIs) are critical for virtually every biological function. Recently, researchers suggested to use supervised learning for the task of classifying pairs of proteins as interacting or not. However, its performance is largely restricted by the availability of truly interacting proteins (labeled). Meanwhile, there exists a considerable amount of protein pairs where an association appears between two partners, but not enough experimental evidence to support it as a direct interaction (partially labeled).
-
Results: We propose a semi-supervised multi-task framework for predicting PPIs from not only labeled, but also partially labeled reference sets. The basic idea is to perform multi-task learning on a supervised classification task and a semi-supervised auxiliary task. The supervised classifier trains a multi-layer perceptron network for PPI predictions from labeled examples. The semi-supervised auxiliary task shares network layers of the supervised classifier and trains with partially labeled examples. Semi-supervision could be utilized in multiple ways. We tried three approaches in this article, (i) classification (to distinguish partial positives with negatives); (ii) ranking (to rate partial positive more likely than negatives); (iii) embedding (to make data clusters get similar labels). We applied this framework to improve the identification of interacting pairs between HIV-1 and human proteins. Our method improved upon the state-of-the-art method for this task indicating the benefits of semi-supervised multi-task learning using auxiliary information.
Citations
@article{qi2010semi,
title={Semi-supervised multi-task learning for predicting interactions between HIV-1 and human proteins},
author={Qi, Yanjun and Tastan, Oznur and Carbonell, Jaime G and Klein-Seetharaman, Judith and Weston, Jason},
journal={Bioinformatics},
volume={26},
number={18},
pages={i645--i652},
year={2010},
publisher={Oxford University Press}
}


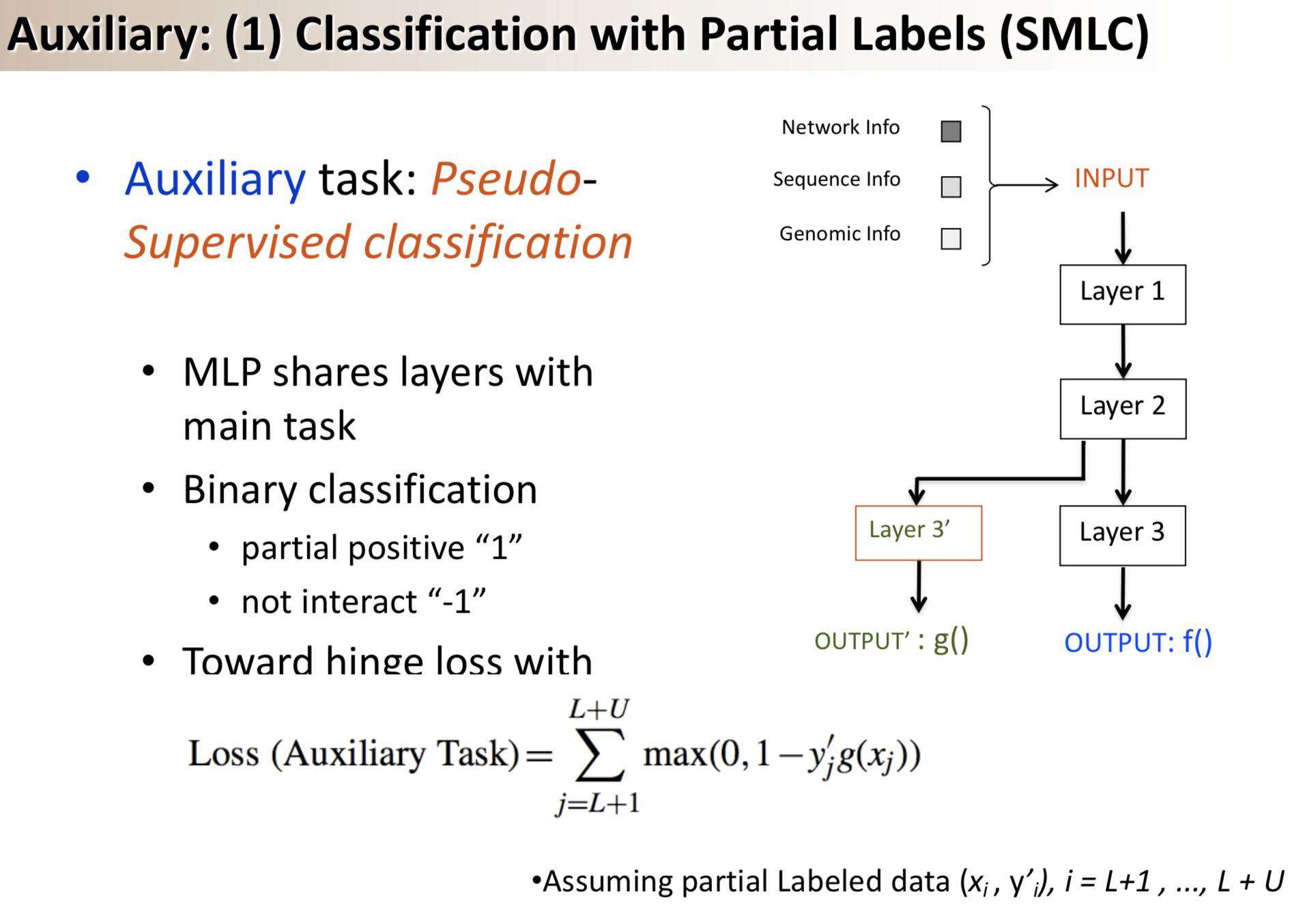
Having trouble with our tools? Please contact Yanjun Qi and we’ll help you sort it out.