Rank-Metric (Index of Posts):
This categoy of tools aims to learn data-driven metric models for the purpose of recommendation or ranking..
This includes:
10 Dec 2017
Prototype Matching Networks : A novel deep learning architecture for Large-Scale Multi-label Genomic Sequence Classification
Abstract
One of the fundamental tasks in understanding genomics is the problem of predicting Transcription Factor Binding Sites (TFBSs). With more than hundreds of Transcription Factors (TFs) as labels, genomic-sequence based TFBS prediction is a challenging multi-label classification task. There are two major biological mechanisms for TF binding: (1) sequence-specific binding patterns on genomes known as “motifs” and (2) interactions among TFs known as co-binding effects. In this paper, we propose a novel deep architecture, the Prototype Matching Network (PMN) to mimic the TF binding mechanisms. Our PMN model automatically extracts prototypes (“motif”-like features) for each TF through a novel prototype-matching loss. Borrowing ideas from few-shot matching models, we use the notion of support set of prototypes and an LSTM to learn how TFs interact and bind to genomic sequences. On a reference TFBS dataset with 2.1 million genomic sequences, PMN significantly outperforms baselines and validates our design choices empirically. To our knowledge, this is the first deep learning architecture that introduces prototype learning and considers TF-TF interactions for large-scale TFBS prediction. Not only is the proposed architecture accurate, but it also models the underlying biology.
Citations
@article{lanchantin2017prototype,
title={Prototype Matching Networks for Large-Scale Multi-label Genomic Sequence Classification},
author={Lanchantin, Jack and Sekhon, Arshdeep and Singh, Ritambhara and Qi, Yanjun},
journal={arXiv preprint arXiv:1710.11238},
year={2017}
}
Having trouble with our tools? Please contact Jack and we’ll help you sort it out.
12 Jun 2017
Abstract
When analyzing the genome, researchers have discovered that proteins bind to DNA based on certain patterns of the DNA sequence known as “motifs”. However, it is difficult to manually construct motifs due to their complexity. Recently, externally learned memory models have proven to be effective methods for reasoning over inputs and supporting sets. In this work, we present memory matching networks (MMN) for classifying DNA sequences as protein binding sites. Our model learns a memory bank of encoded motifs, which are dynamic memory modules, and then matches a new test sequence to each of the motifs to classify the sequence as a binding or nonbinding site.
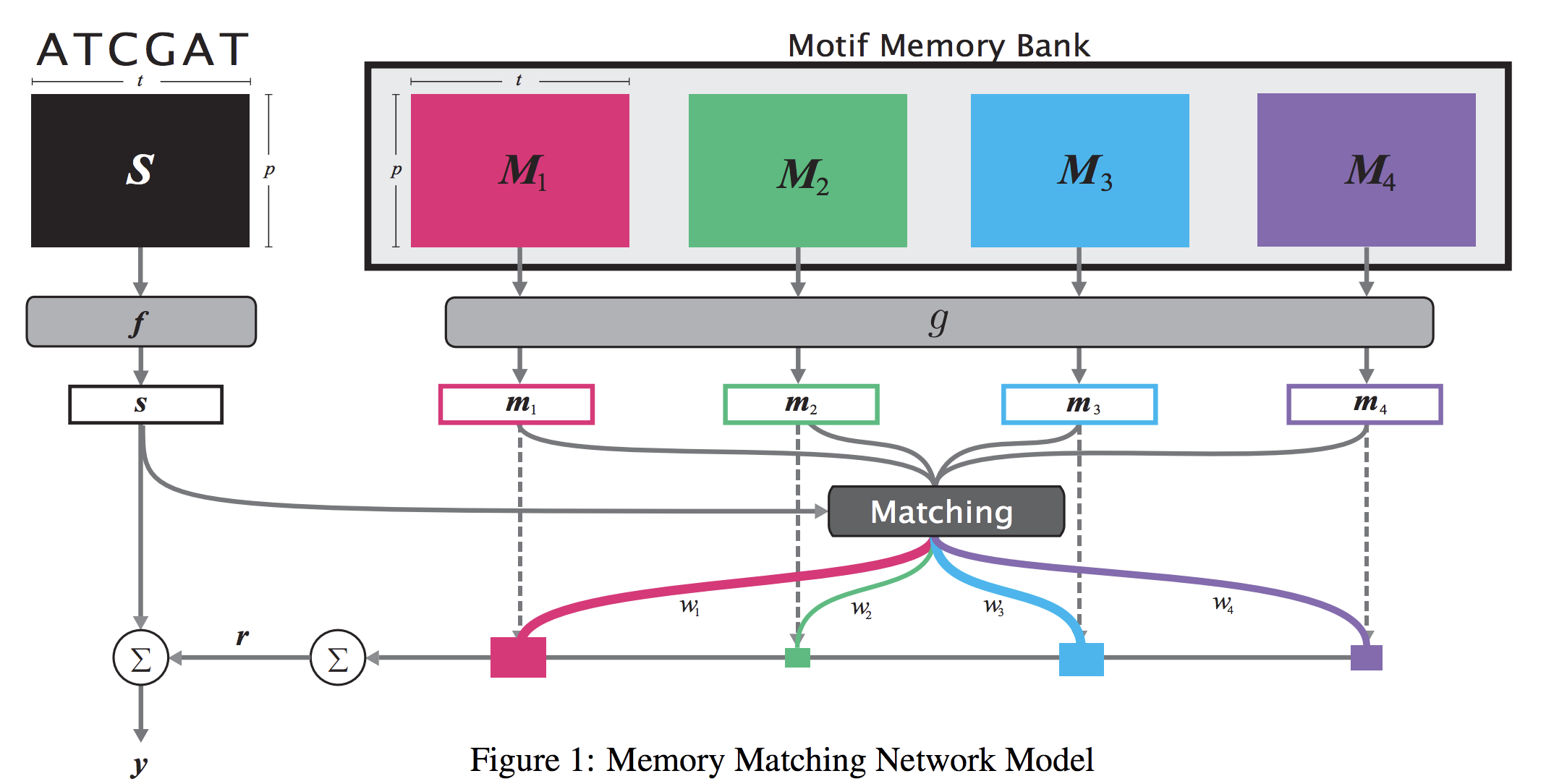
Citations
@article{lanchantin2017memory,
title={Memory Matching Networks for Genomic Sequence Classification},
author={Lanchantin, Jack and Singh, Ritambhara and Qi, Yanjun},
journal={arXiv preprint arXiv:1702.06760},
year={2017}
}
Having trouble with our tools? Please contact Jack and we’ll help you sort it out.
01 May 2011
Paper0: Learning to rank with (a lot of) word features
-
PDF
-
Abstract
In this article we present Supervised Semantic Indexing which defines a class of nonlinear (quadratic) models that are discriminatively trained to directly map from the word content in a query-document or document-document pair to a ranking score. Like Latent Semantic Indexing (LSI), our models take account of correlations between words (synonymy, polysemy). However, unlike LSI our models are trained from a supervised signal directly on the ranking task of interest, which we argue is the reason for our superior results. As the query and target texts are modeled separately, our approach is easily generalized to different retrieval tasks, such as cross-language retrieval or online advertising placement. Dealing with models on all pairs of words features is computationally challenging. We propose several improvements to our basic model for addressing this issue, including low rank (but diagonal preserving) representations, correlated feature hashing and sparsification. We provide an empirical study of all these methods on retrieval tasks based on Wikipedia documents as well as an Internet advertisement task. We obtain state-of-the-art performance while providing realistically scalable methods.
Paper1: Polynomial semantic indexing
- PDF
- Bing Bai, Jason Weston, David Grangier, Ronan Collobert, Kunihiko Sadamasa, Yanjun Qi, Corinna Cortes, Mehryar Mohri
- 2009 Conference on Advances in Neural Information Processing Systems
- Abstract
We present a class of nonlinear (polynomial) models that are discriminatively trained to directly map from the word content in a query-document or document-document pair to a ranking score. Dealing with polynomial models on word features is computationally challenging. We propose a low rank (but diagonal preserving) representation of our polynomial models to induce feasible memory and computation requirements. We provide an empirical study on retrieval tasks based on Wikipedia documents, where we obtain state-of-the-art performance while providing realistically scalable methods.
Paper2: Retrieving Medical Records with “sennamed”: NEC Labs America at TREC 2012 Medical Records Track
-
PDF
-
Abstract
In this notebook, we describe the automatic retrieval runs from NEC Laboratories America (NECLA) for the Text REtrieval Conference (TREC) 2012 Medical Records track. Our approach is based on a combination of UMLS medical concept detection and a set of simple retrieval models. Our best run, sennamed2, has achieved the best inferred average precision (infAP) score on 5 of the 47 test topics, and obtained a higher score than the median of all submission runs on 27 other topics. Overall, sennamed2 ranks at the second place amongst all the 82 automatic runs submitted for this track, and obtains the third place amongst both automatic and manual submissions.
-
PDF
-
Abstract
With the advancement of genome-wide monitoring technologies, molecular expression data have become widely used for diagnosing cancer through tumor or blood samples. When mining molecular signature data, the process of comparing samples through an adaptive distance function is fundamental but difficult, as such datasets are normally heterogeneous and high dimensional. In this article, we present kernelized information-theoretic metric learning (KITML) algorithms that optimize a distance function to tackle the cancer diagnosis problem and scale to high dimensionality. By learning a nonlinear transformation in the input space implicitly through kernelization, KITML permits efficient optimization, low storage, and improved learning of distance metric. We propose two novel applications of KITML for diagnosing cancer using high-dimensional molecular profiling data: (1) for sample-level cancer diagnosis, the learned metric is used to improve the performance of k-nearest neighbor classification; and (2) for estimating the severity level or stage of a group of samples, we propose a novel set-based ranking approach to extend KITML. For the sample-level cancer classification task, we have evaluated on 14 cancer gene microarray datasets and compared with eight other state-of-the-art approaches. The results show that our approach achieves the best overall performance for the task of molecular-expression-driven cancer sample diagnosis. For the group-level cancer stage estimation, we test the proposed set-KITML approach using three multi-stage cancer microarray datasets, and correctly estimated the stages of sample groups for all three studies.
Paper4: Learning preferences with millions of parameters by enforcing sparsity
- PDF
-
Talk
- Abstract
We study the retrieval task that ranks a set of objects for a given query in the pair wise preference learning framework. Recently researchers found out that raw features (e.g. words for text retrieval) and their pair wise features which describe relationships between two raw features (e.g. word synonymy or polysemy) could greatly improve the retrieval precision. However, most existing methods can not scale up to problems with many raw features (e.g. English vocabulary), due to the prohibitive computational cost on learning and the memory requirement to store a quadratic number of parameters. In this paper, we propose to learn a sparse representation of the pair wise features under the preference learning framework using the L1 regularization. Based on stochastic gradient descent, an online algorithm is devised to enforce the sparsity using a mini-batch shrinkage strategy. On multiple benchmark datasets, we show that our method achieves better performance with fast convergence, and takes much less memory on models with millions of parameters.
Citations
@techreport{qi2012retrieving,
title={Retrieving medical records with sennamed: Nec labs america at trec 2012 medical records track},
author={Qi, Yanjun and Laquerre, Pierre-Fran{\c{c}}ois},
year={2012},
institution={NEC Laboratories America Inc Princeton NJ}
}
Having trouble with our tools? Please contact Yanjun Qi and we’ll help you sort it out.