Tagging-Genome (Index of Posts):
This categoy of tools aims to tag local genome pieces into functional types..
This includes:
01 Jun 2020
Title: FastSK: Fast Sequence Analysis with Gapped String Kernels
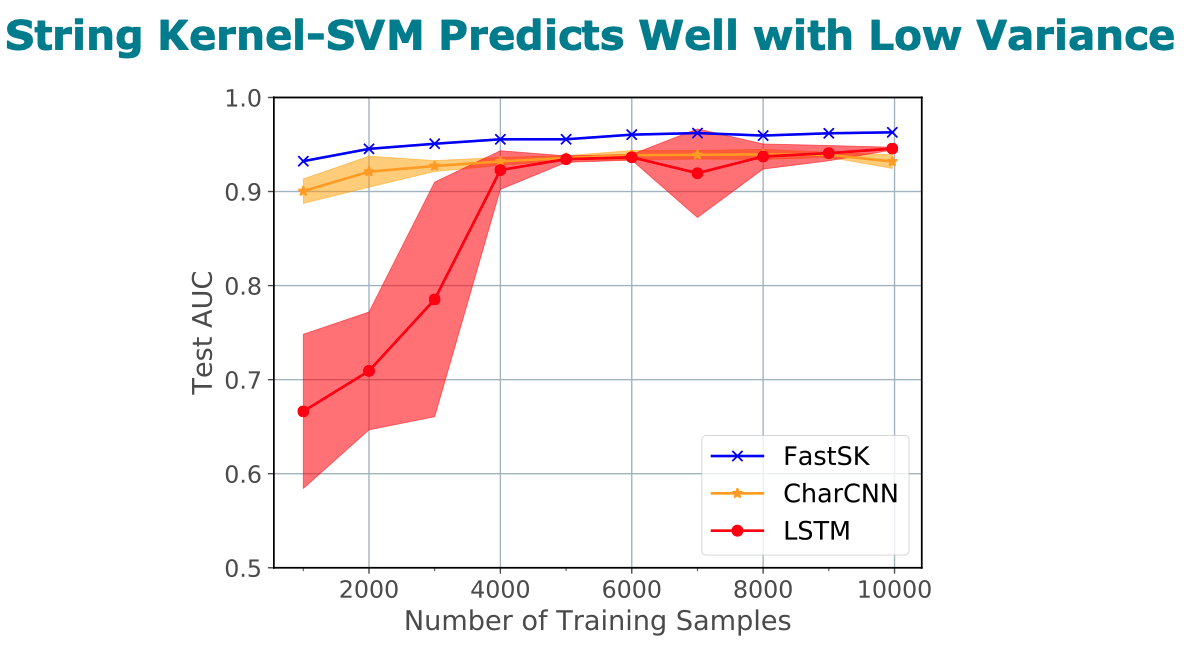

Abstract
Gapped k-mer kernels with Support Vector Machines (gkm-SVMs)
have achieved strong predictive performance on regulatory DNA sequences
on modestly-sized training sets. However, existing gkm-SVM algorithms
suffer from the slow kernel computation time, as they depend
exponentially on the sub-sequence feature-length, number of mismatch
positions, and the task’s alphabet size.
In this work, we introduce a fast and scalable algorithm for
calculating gapped k-mer string kernels. Our method, named FastSK,
uses a simplified kernel formulation that decomposes the kernel
calculation into a set of independent counting operations over the
possible mismatch positions. This simplified decomposition allows us
to devise a fast Monte Carlo approximation that rapidly converges.
FastSK can scale to much greater feature lengths, allows us to
consider more mismatches, and is performant on a variety of sequence
analysis tasks. On 10 DNA transcription factor binding site (TFBS)
prediction datasets, FastSK consistently matches or outperforms the
state-of-the-art gkmSVM-2.0 algorithms in AUC, while achieving
average speedups in kernel computation of 100 times and speedups of
800 times for large feature lengths. We further show that FastSK
outperforms character-level recurrent and convolutional neural
networks across all 10 TFBS tasks. We then extend FastSK to 7
English medical named entity recognition datasets and 10 protein
remote homology detection datasets. FastSK consistently matches or
outperforms these baselines.
Our algorithm is available as a Python package and as C++ source code.
(Available for download at https://github.com/Qdata/FastSK/.
Install with the command make or pip install)
Citations
@article{10.1093/bioinformatics/btaa817,
author = {Blakely, Derrick and Collins, Eamon and Singh, Ritambhara and Norton, Andrew and Lanchantin, Jack and Qi, Yanjun},
title = "{FastSK: fast sequence analysis with gapped string kernels}",
journal = {Bioinformatics},
volume = {36},
number = {Supplement_2},
pages = {i857-i865},
year = {2020},
month = {12},
issn = {1367-4803},
doi = {10.1093/bioinformatics/btaa817},
url = {https://doi.org/10.1093/bioinformatics/btaa817},
eprint = {https://academic.oup.com/bioinformatics/article-pdf/36/Supplement\_2/i857/35337038/btaa817.pdf},
}
Having trouble with our tools? Please contact Yanjun Qi and we’ll help you sort it out.
01 May 2020
Title: Graph Convolutional Networks for Epigenetic State Prediction Using Both Sequence and 3D Genome Data
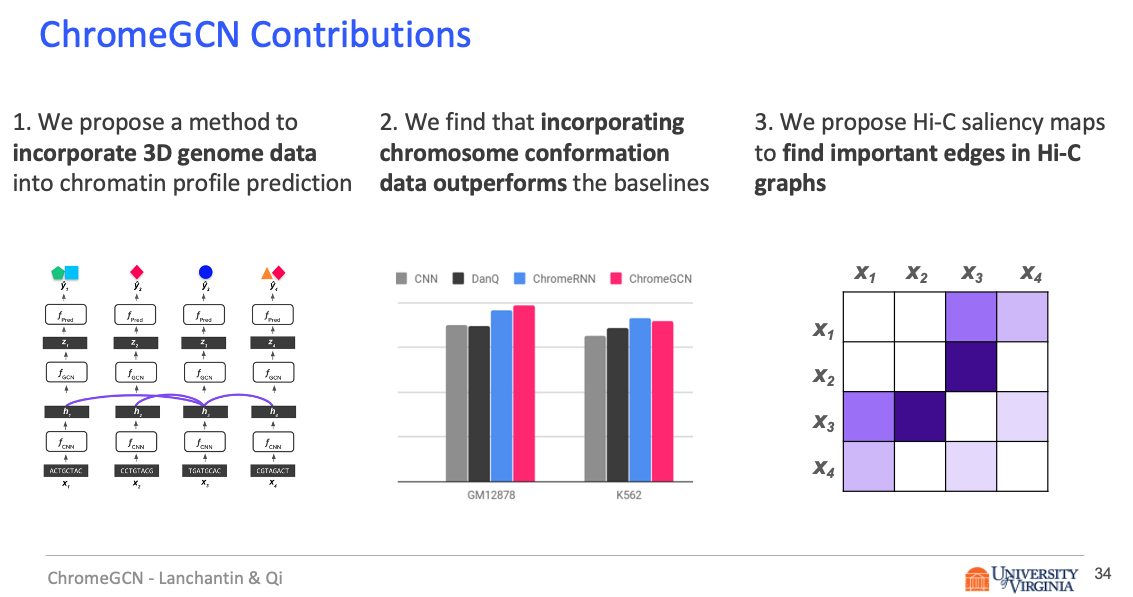
Abstract
Motivation
Predictive models of DNA chromatin profile (i.e. epigenetic state), such as transcription factor binding, are essential for understanding regulatory processes and developing gene therapies. It is known that the 3D genome, or spatial structure of DNA, is highly influential in the chromatin profile. Deep neural networks have achieved state of the art performance on chromatin profile prediction by using short windows of DNA sequences independently. These methods, however, ignore the long-range dependencies when predicting the chromatin profiles because modeling the 3D genome is challenging.
Results
In this work, we introduce ChromeGCN, a graph convolutional network for chromatin profile prediction by fusing both local sequence and long-range 3D genome information. By incorporating the 3D genome, we relax the independent and identically distributed assumption of local windows for a better representation of DNA. ChromeGCN explicitly incorporates known long-range interactions into the modeling, allowing us to identify and interpret those important long-range dependencies in influencing chromatin profiles. We show experimentally that by fusing sequential and 3D genome data using ChromeGCN, we get a significant improvement over the state-of-the-art deep learning methods as indicated by three metrics. Importantly, we show that ChromeGCN is particularly useful for identifying epigenetic effects in those DNA windows that have a high degree of interactions with other DNA windows.
Citations
@article{10.1093/bioinformatics/btaa793,
author = {Lanchantin, Jack and Qi, Yanjun},
title = "{Graph convolutional networks for epigenetic state prediction using both sequence and 3D genome data}",
journal = {Bioinformatics},
volume = {36},
number = {Supplement_2},
pages = {i659-i667},
year = {2020},
month = {12},
issn = {1367-4803},
doi = {10.1093/bioinformatics/btaa793},
url = {https://doi.org/10.1093/bioinformatics/btaa793},
eprint = {https://academic.oup.com/bioinformatics/article-pdf/36/Supplement\_2/i659/35336695/btaa793.pdf},
}
Having trouble with our tools? Please contact Jack and we’ll help you sort it out.
10 Dec 2017
Prototype Matching Networks : A novel deep learning architecture for Large-Scale Multi-label Genomic Sequence Classification
Abstract
One of the fundamental tasks in understanding genomics is the problem of predicting Transcription Factor Binding Sites (TFBSs). With more than hundreds of Transcription Factors (TFs) as labels, genomic-sequence based TFBS prediction is a challenging multi-label classification task. There are two major biological mechanisms for TF binding: (1) sequence-specific binding patterns on genomes known as “motifs” and (2) interactions among TFs known as co-binding effects. In this paper, we propose a novel deep architecture, the Prototype Matching Network (PMN) to mimic the TF binding mechanisms. Our PMN model automatically extracts prototypes (“motif”-like features) for each TF through a novel prototype-matching loss. Borrowing ideas from few-shot matching models, we use the notion of support set of prototypes and an LSTM to learn how TFs interact and bind to genomic sequences. On a reference TFBS dataset with 2.1 million genomic sequences, PMN significantly outperforms baselines and validates our design choices empirically. To our knowledge, this is the first deep learning architecture that introduces prototype learning and considers TF-TF interactions for large-scale TFBS prediction. Not only is the proposed architecture accurate, but it also models the underlying biology.
Citations
@article{lanchantin2017prototype,
title={Prototype Matching Networks for Large-Scale Multi-label Genomic Sequence Classification},
author={Lanchantin, Jack and Sekhon, Arshdeep and Singh, Ritambhara and Qi, Yanjun},
journal={arXiv preprint arXiv:1710.11238},
year={2017}
}
Having trouble with our tools? Please contact Jack and we’ll help you sort it out.
30 Jul 2017
Paper: @Arxiv + Published at [NIPS2017]
(https://papers.nips.cc/paper/7255-attend-and-predict-understanding-gene-regulation-by-selective-attention-on-chromatin.pdf)
Abstract:
The past decade has seen a revolution in genomic technologies that enable a flood of genome-wide profiling of chromatin marks. Recent literature tried to understand gene regulation by predicting gene expression from large-scale chromatin measurements. Two fundamental challenges exist for such learning tasks: (1) genome-wide chromatin signals are spatially structured, high-dimensional and highly modular; and (2) the core aim is to understand what are the relevant factors and how they work together? Previous studies either failed to model complex dependencies among input signals or relied on separate feature analysis to explain the decisions. This paper presents an attention-based deep learning approach; we call AttentiveChrome, that uses a unified architecture to model and to interpret dependencies among chromatin factors for controlling gene regulation. AttentiveChrome uses a hierarchy of multiple Long short-term memory (LSTM) modules to encode the input signals and to model how various chromatin marks cooperate automatically. AttentiveChrome trains two levels of attention jointly with the target prediction, enabling it to attend differentially to relevant marks and to locate important positions per mark. We evaluate the model across 56 different cell types (tasks) in human. Not only is the proposed architecture more accurate, but its attention scores also provide a better interpretation than state-of-the-art feature visualization methods such as saliency map.
Code and data are shared at www.deepchrome.org
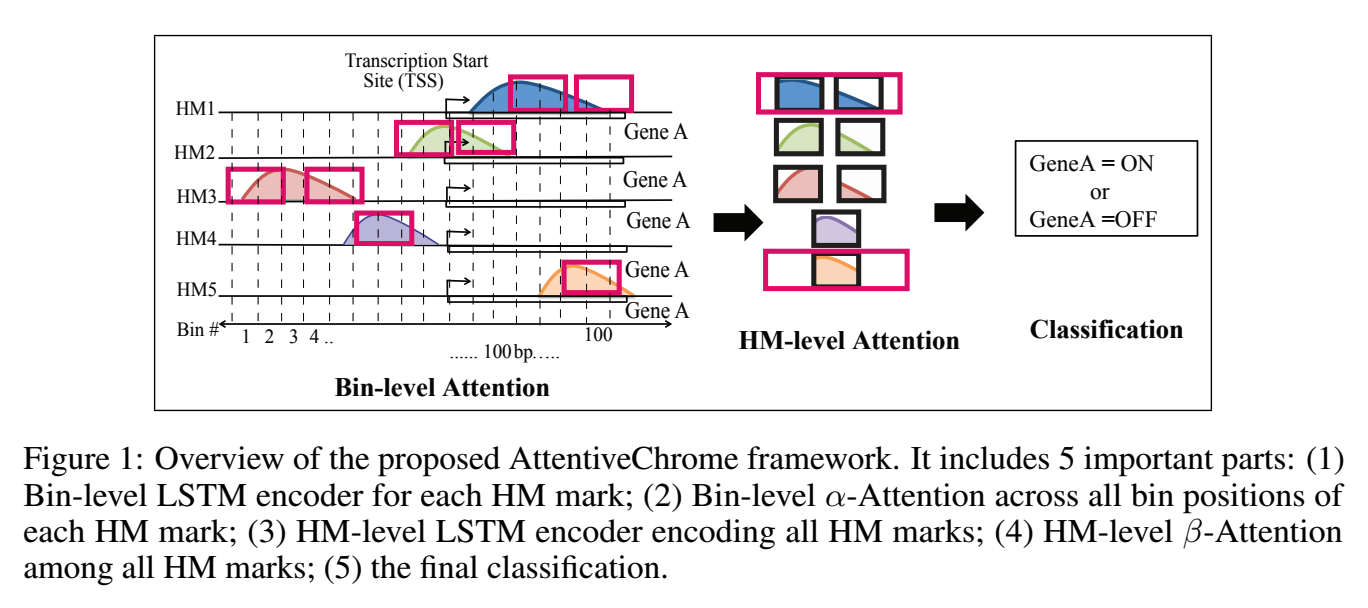
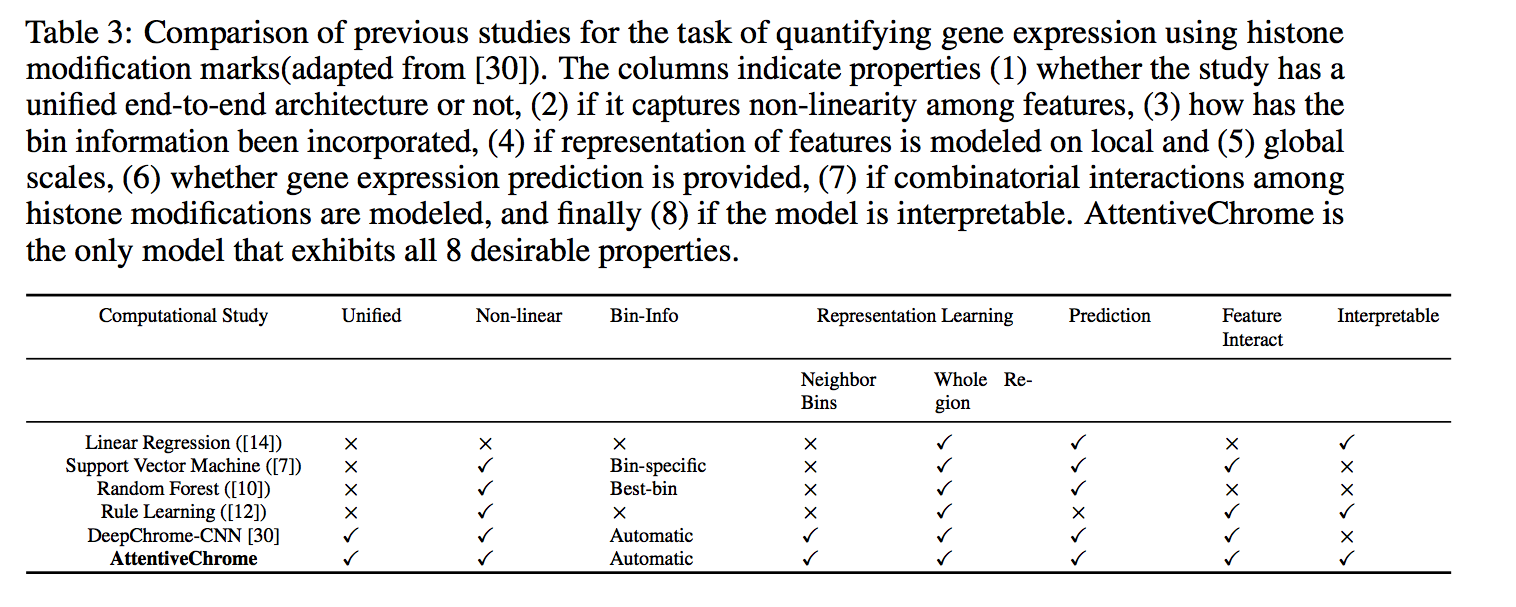
Citations
@inproceedings{singh2017attend,
title={Attend and Predict: Understanding Gene Regulation by Selective Attention on Chromatin},
author={Singh, Ritambhara and Lanchantin, Jack and Sekhon, Arshdeep and Qi, Yanjun},
booktitle={Advances in Neural Information Processing Systems},
pages={6769--6779},
year={2017}
}
Having trouble with our tools? Please contact Rita and we’ll help you sort it out.
12 Jun 2017
Abstract
When analyzing the genome, researchers have discovered that proteins bind to DNA based on certain patterns of the DNA sequence known as “motifs”. However, it is difficult to manually construct motifs due to their complexity. Recently, externally learned memory models have proven to be effective methods for reasoning over inputs and supporting sets. In this work, we present memory matching networks (MMN) for classifying DNA sequences as protein binding sites. Our model learns a memory bank of encoded motifs, which are dynamic memory modules, and then matches a new test sequence to each of the motifs to classify the sequence as a binding or nonbinding site.
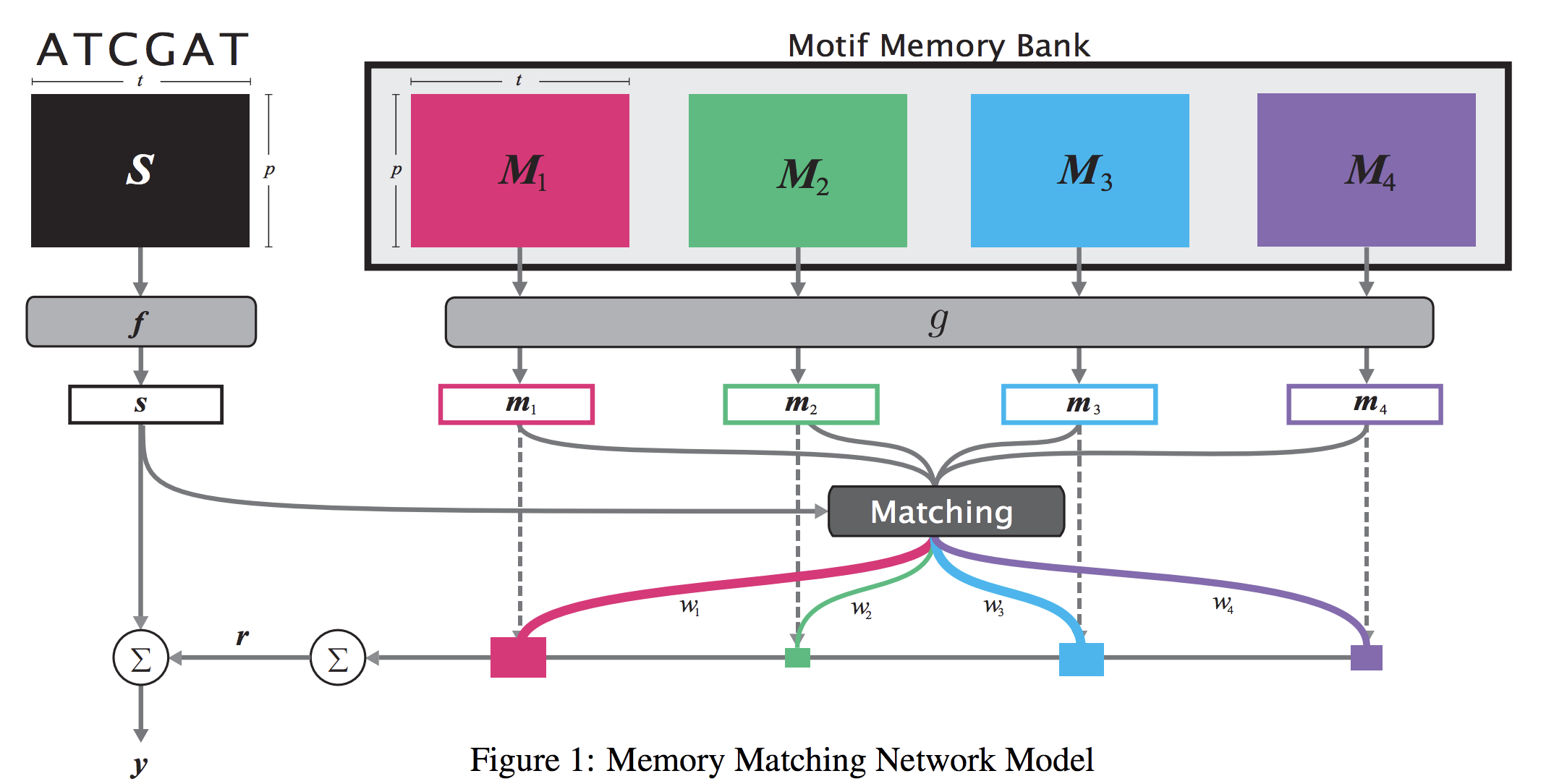
Citations
@article{lanchantin2017memory,
title={Memory Matching Networks for Genomic Sequence Classification},
author={Lanchantin, Jack and Singh, Ritambhara and Qi, Yanjun},
journal={arXiv preprint arXiv:1702.06760},
year={2017}
}
Having trouble with our tools? Please contact Jack and we’ll help you sort it out.
11 Jun 2017
Abstract:
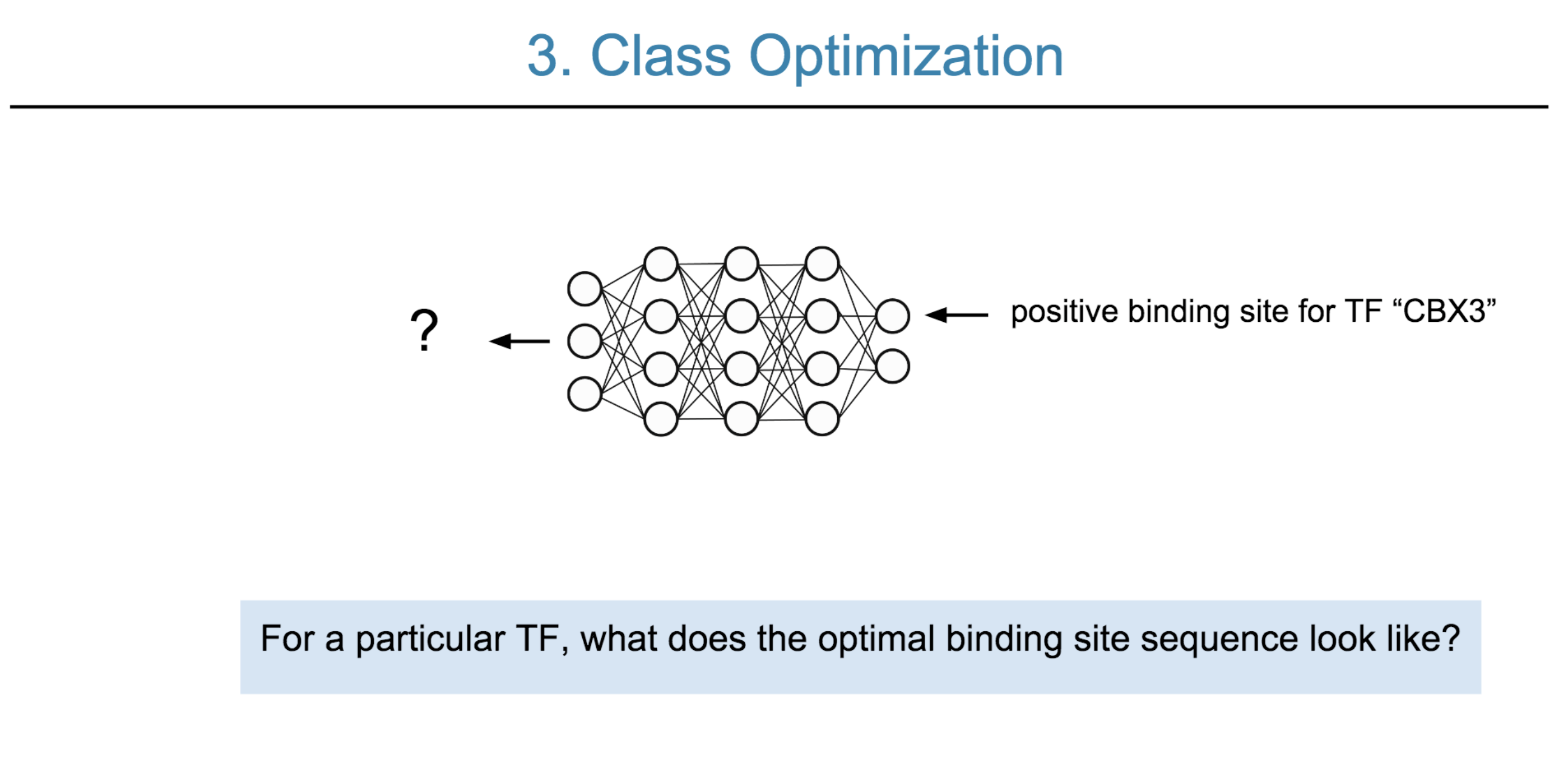
Deep neural network (DNN) models have recently obtained state-of-the-art prediction accuracy for the transcription factor binding (TFBS) site classification task. However, it remains unclear how these approaches identify meaningful DNA sequence signals and give insights as to why TFs bind to certain locations. In this paper, we propose a toolkit called the Deep Motif Dashboard (DeMo Dashboard) which provides a suite of visualization strategies to extract motifs, or sequence patterns from deep neural network models for TFBS classification. We demonstrate how to visualize and understand three important DNN models: convolutional, recurrent, and convolutional-recurrent networks. Our first visualization method is finding a test sequence’s saliency map which uses first-order derivatives to describe the importance of each nucleotide in making the final prediction. Second, considering recurrent models make predictions in a temporal manner (from one end of a TFBS sequence to the other), we introduce temporal output scores, indicating the prediction score of a model over time for a sequential input. Lastly, a class-specific visualization strategy finds the optimal input sequence for a given TFBS positive class via stochastic gradient optimization. Our experimental results indicate that a convolutional-recurrent architecture performs the best among the three architectures. The visualization techniques indicate that CNN-RNN makes predictions by modeling both motifs as well as dependencies among them.
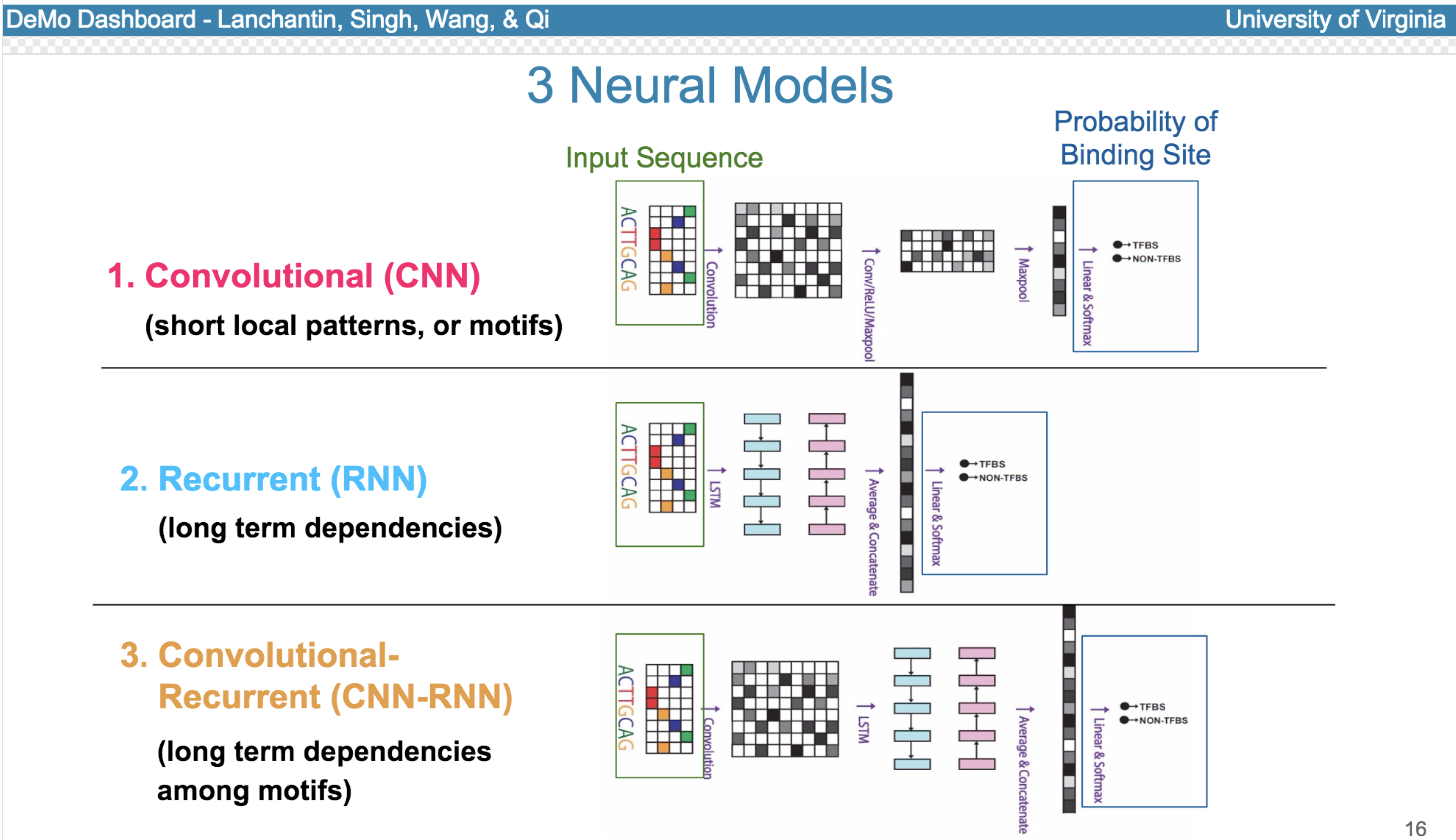
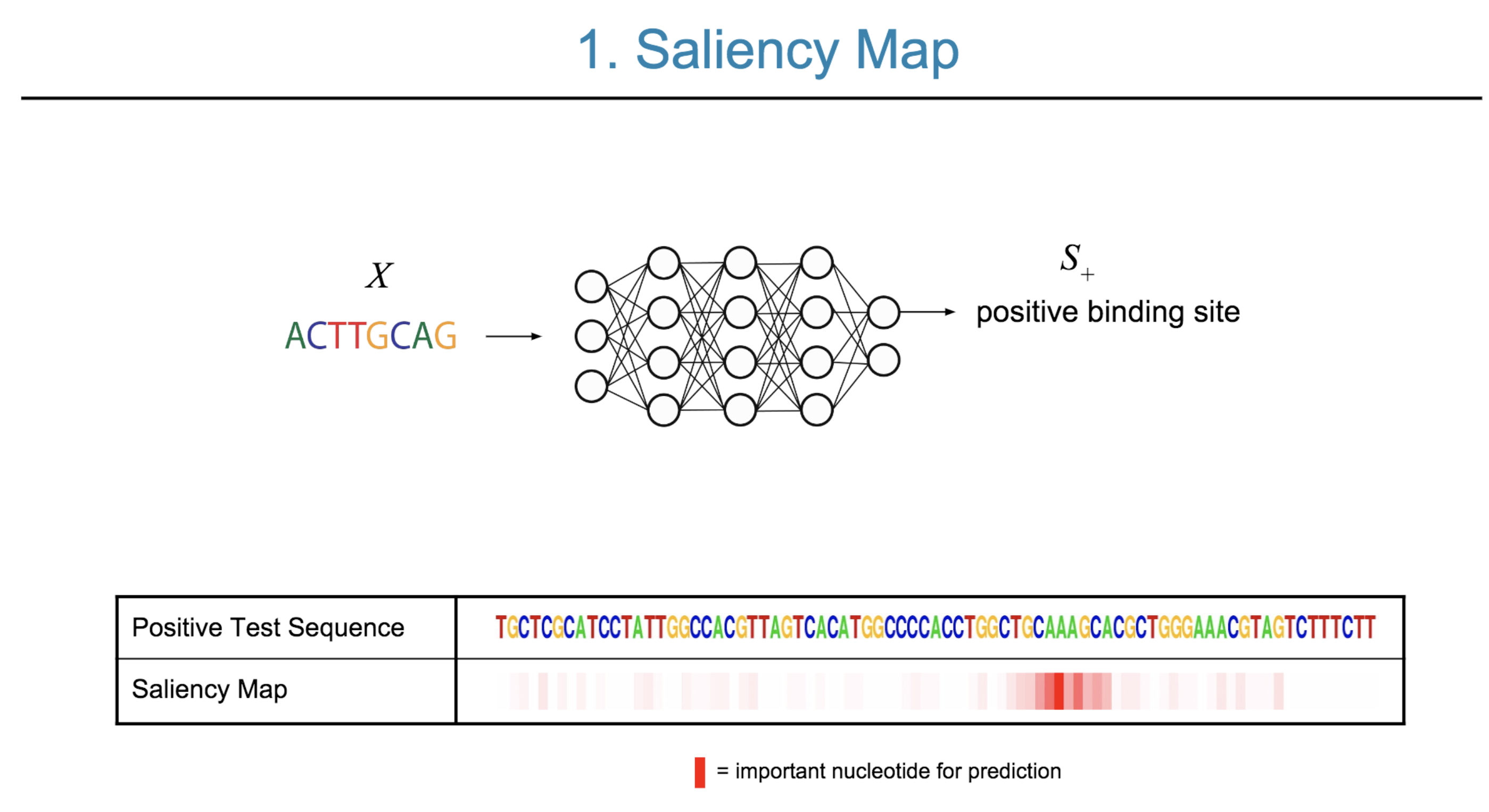
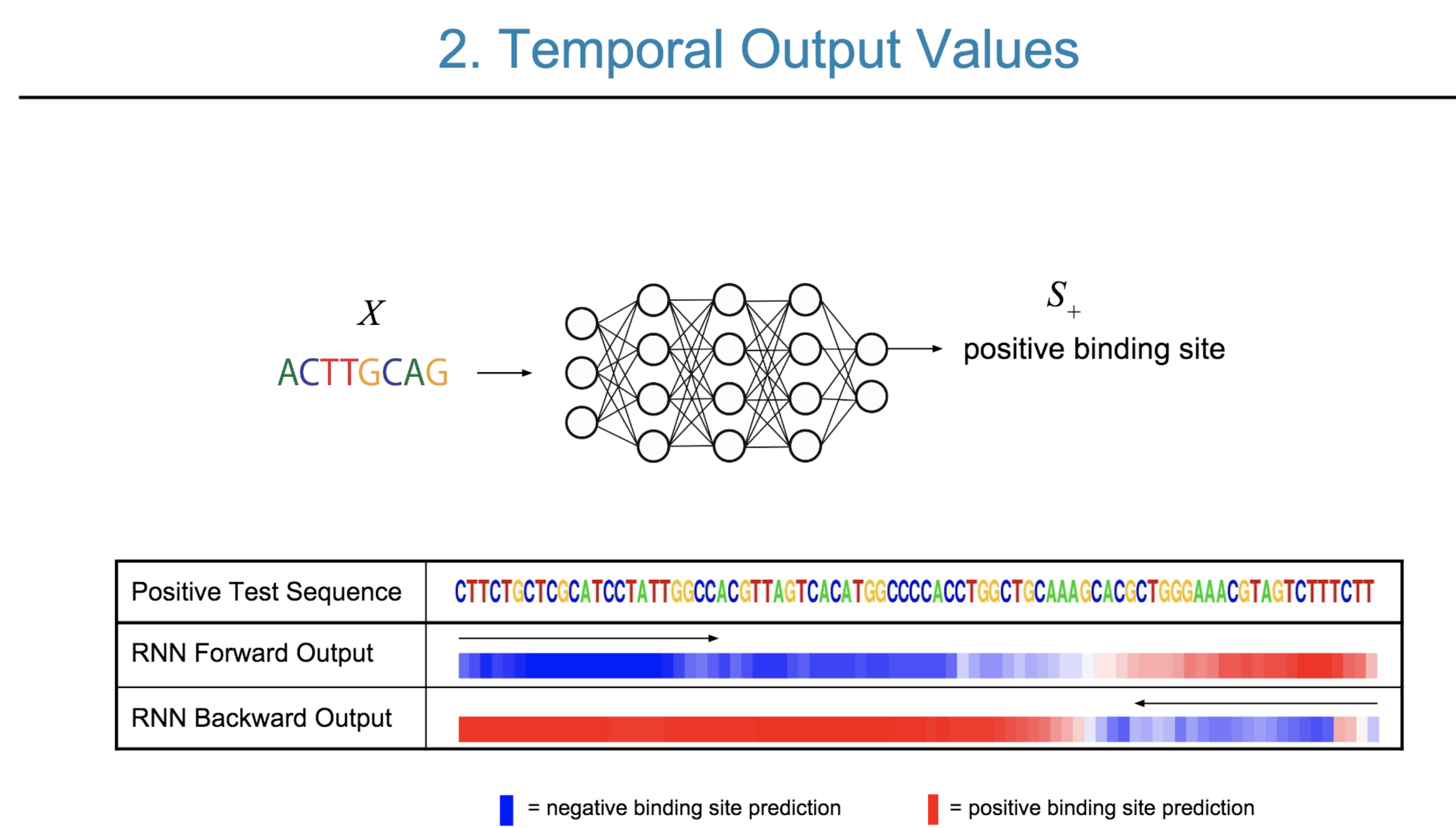
Citations
@inproceedings{lanchantin2017deep,
title={Deep motif dashboard: Visualizing and understanding genomic sequences using deep neural networks},
author={Lanchantin, Jack and Singh, Ritambhara and Wang, Beilun and Qi, Yanjun},
booktitle={PACIFIC SYMPOSIUM ON BIOCOMPUTING 2017},
pages={254--265},
year={2017},
organization={World Scientific}
}
Having trouble with our tools? Please contact Jack and we’ll help you sort it out.
20 Jun 2016
Abstract:
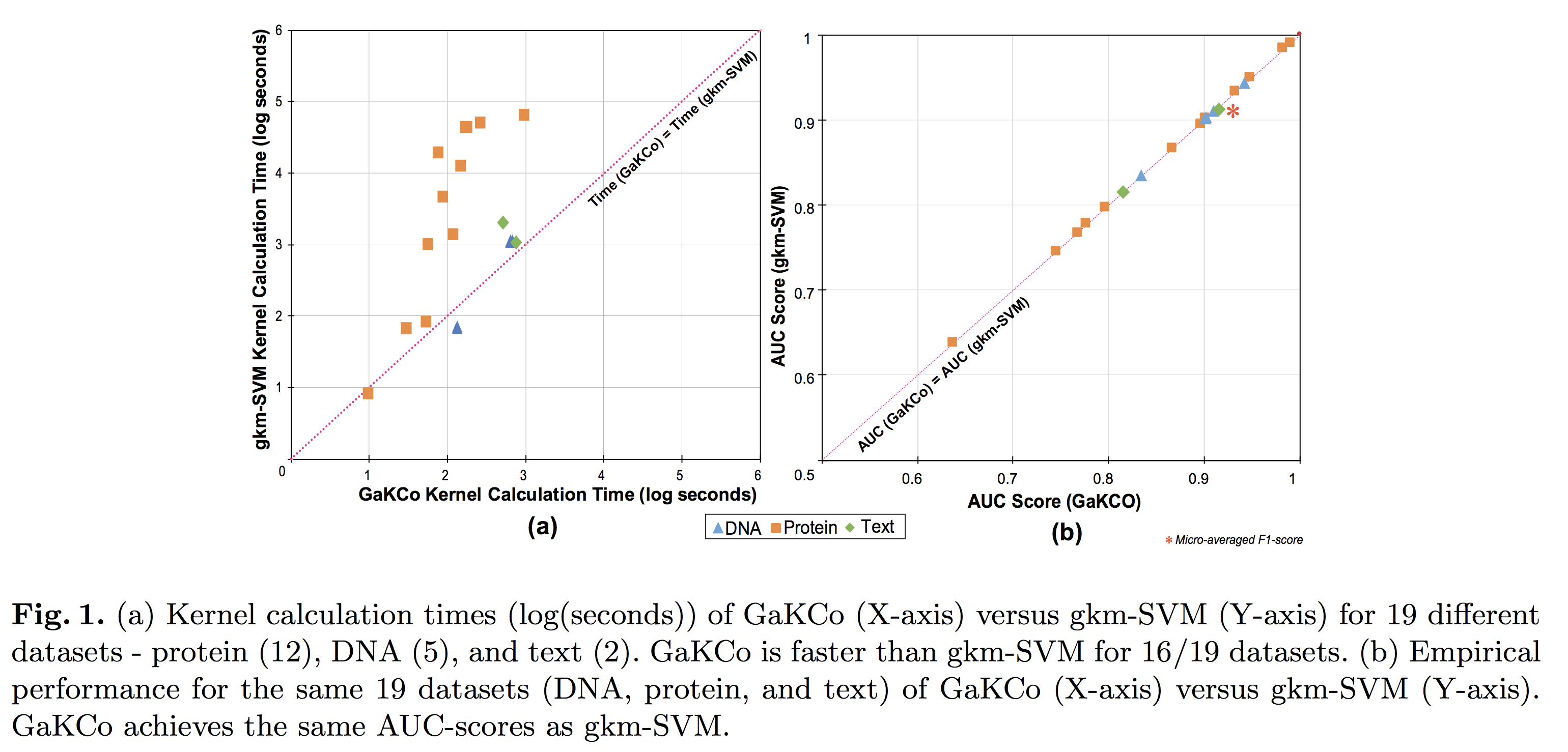
String Kernel (SK) techniques, especially those using gapped k-mers as features (gk), have obtained great success in classifying sequences like DNA, protein, and text. However, the state-of-the-art gk-SK runs extremely slow when we increase the dictionary size (Σ) or allow more mismatches (M). This is because current gk-SK uses a trie-based algorithm to calculate co-occurrence of mismatched substrings resulting in a time cost proportional to O(ΣM). We propose a \textbf{fast} algorithm for calculating \underline{Ga}pped k-mer \underline{K}ernel using \underline{Co}unting (GaKCo). GaKCo uses associative arrays to calculate the co-occurrence of substrings using cumulative counting. This algorithm is fast, scalable to larger Σ and M, and naturally parallelizable. We provide a rigorous asymptotic analysis that compares GaKCo with the state-of-the-art gk-SK. Theoretically, the time cost of GaKCo is independent of the ΣM term that slows down the trie-based approach. Experimentally, we observe that GaKCo achieves the same accuracy as the state-of-the-art and outperforms its speed by factors of 2, 100, and 4, on classifying sequences of DNA (5 datasets), protein (12 datasets), and character-based English text (2 datasets), respectively.
Citations
@inproceedings{singh_gakco:_2017,
location = {Cham},
title = {GaKCo: A Fast Gapped k-mer String Kernel Using Counting},
isbn = {978-3-319-71249-9},
pages = {356--373},
booktitle = {Machine Learning and Knowledge Discovery in Databases},
publisher = {Springer International Publishing},
author = {Singh, Ritambhara and Sekhon, Arshdeep and Kowsari, Kamran and Lanchantin, Jack and Wang, Beilun and Qi, Yanjun},
editor = {Ceci, Michelangelo and Hollmén, Jaakko and Todorovski, Ljupčo and Vens, Celine and Džeroski, Sašo},
date = {2017}
}
Having trouble with our tools? Please contact Rita and we’ll help you sort it out.