Predict-Graph (Index of Posts):
This categoy of tools aims to discover dependency graphs among interested variables..
This includes:
11 Nov 2020
- authors: Jack Lanchantin, Arshdeep Sekhon, Clint Miller, Yanjun Qi
Abstract
The novel coronavirus SARS-CoV-2, which causes Coronavirus disease 2019 (COVID-19), is a significant threat to worldwide public health. Viruses such as SARS-CoV-2 infect the human body by forming interactions between virus proteins and human proteins that compromise normal human protein-protein interactions (PPI). Current in vivo methods to identify PPIs between a novel virus and humans are slow, costly, and difficult to cover the vast interaction space. We propose a novel deep learning architecture designed for in silico PPI prediction and a transfer learning approach to predict interactions between novel virus proteins and human proteins. We show that our approach outperforms the state-of-the-art methods significantly in predicting Virus–Human protein interactions for SARS-CoV-2, H1N1, and Ebola.
Citations
@article {Lanchantin2020.12.14.422772,
author = {Lanchantin, Jack and Sekhon, Arshdeep and Miller, Clint and Qi, Yanjun},
title = {Transfer Learning with MotifTransformers for Predicting Protein-Protein Interactions Between a Novel Virus and Humans},
elocation-id = {2020.12.14.422772},
year = {2020},
doi = {10.1101/2020.12.14.422772},
publisher = {Cold Spring Harbor Laboratory},
URL = {https://www.biorxiv.org/content/early/2020/12/15/2020.12.14.422772},
eprint = {https://www.biorxiv.org/content/early/2020/12/15/2020.12.14.422772.full.pdf},
journal = {bioRxiv}
}
Having trouble with our tools? Please contact Jack and we’ll help you sort it out.
01 May 2020
Title: Graph Convolutional Networks for Epigenetic State Prediction Using Both Sequence and 3D Genome Data
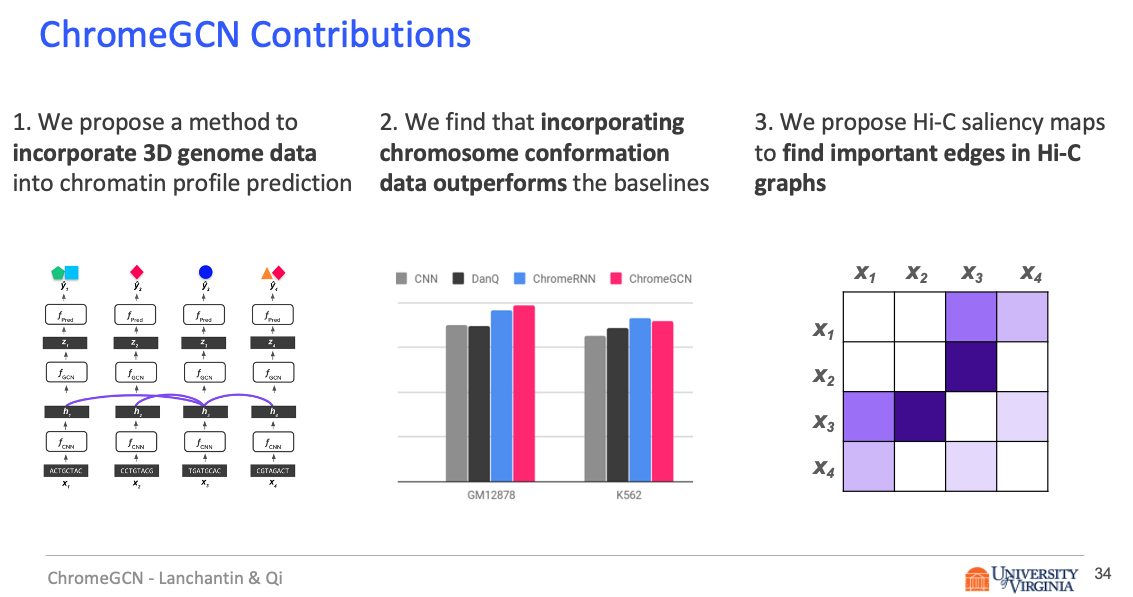
Abstract
Motivation
Predictive models of DNA chromatin profile (i.e. epigenetic state), such as transcription factor binding, are essential for understanding regulatory processes and developing gene therapies. It is known that the 3D genome, or spatial structure of DNA, is highly influential in the chromatin profile. Deep neural networks have achieved state of the art performance on chromatin profile prediction by using short windows of DNA sequences independently. These methods, however, ignore the long-range dependencies when predicting the chromatin profiles because modeling the 3D genome is challenging.
Results
In this work, we introduce ChromeGCN, a graph convolutional network for chromatin profile prediction by fusing both local sequence and long-range 3D genome information. By incorporating the 3D genome, we relax the independent and identically distributed assumption of local windows for a better representation of DNA. ChromeGCN explicitly incorporates known long-range interactions into the modeling, allowing us to identify and interpret those important long-range dependencies in influencing chromatin profiles. We show experimentally that by fusing sequential and 3D genome data using ChromeGCN, we get a significant improvement over the state-of-the-art deep learning methods as indicated by three metrics. Importantly, we show that ChromeGCN is particularly useful for identifying epigenetic effects in those DNA windows that have a high degree of interactions with other DNA windows.
Citations
@article{10.1093/bioinformatics/btaa793,
author = {Lanchantin, Jack and Qi, Yanjun},
title = "{Graph convolutional networks for epigenetic state prediction using both sequence and 3D genome data}",
journal = {Bioinformatics},
volume = {36},
number = {Supplement_2},
pages = {i659-i667},
year = {2020},
month = {12},
issn = {1367-4803},
doi = {10.1093/bioinformatics/btaa793},
url = {https://doi.org/10.1093/bioinformatics/btaa793},
eprint = {https://academic.oup.com/bioinformatics/article-pdf/36/Supplement\_2/i659/35336695/btaa793.pdf},
}
Having trouble with our tools? Please contact Jack and we’ll help you sort it out.
01 Mar 2019
Title: Neural Message Passing for Multi-Label Classification
Abstract
Multi-label classification (MLC) is the task of assigning a set of target labels for a given sample. Modeling the combinatorial label interactions in MLC has been a long-haul challenge. Recurrent neural network (RNN) based encoder-decoder models have shown state-of-the-art performance for solving MLC. However, the sequential nature of modeling label dependencies through an RNN limits its ability in parallel computation, predicting dense labels, and providing interpretable results. In this paper, we propose Message Passing Encoder-Decoder (MPED) Networks, aiming to provide fast, accurate, and interpretable MLC. MPED networks model the joint prediction of labels by replacing all RNNs in the encoder-decoder architecture with message passing mechanisms and dispense with autoregressive inference entirely. The proposed models are simple, fast, accurate, interpretable, and structure-agnostic (can be used on known or unknown structured data). Experiments on seven real-world MLC datasets show the proposed models outperform autoregressive RNN models across five different metrics with a significant speedup during training and testing time.
Citations
@article{lanchantin2018neural,
title={Neural Message Passing for Multi-Label Classification},
author={Lanchantin, Jack and Sekhon, Arshdeep and Qi, Yanjun},
year={2018}
}
Having trouble with our tools? Please contact Jack Lanchantin and we’ll help you sort it out.
20 Jun 2014
- Yunlong He, Yanjun Qi, Koray Kavukcuoglu, Haesun Park
Abstract:
In this paper, we study latent factor models with the dependency structure in the latent space. We propose a general learning framework which induces sparsity on the undirected graphical model imposed on the vector of latent factors. A novel latent factor model SLFA is then proposed as a matrix factorization problem with a special regularization term that encourages collaborative reconstruction. The main benefit (novelty) of the model is that we can simultaneously learn the lower-dimensional representation for data and model the pairwise relationships between latent factors explicitly. An on-line learning algorithm is devised to make the model feasible for large-scale learning problems. Experimental results on two synthetic data and two real-world data sets demonstrate that pairwise relationships and latent factors learned by our model provide a more structured way of exploring high-dimensional data, and the learned representations achieve the state-of-the-art classification performance.
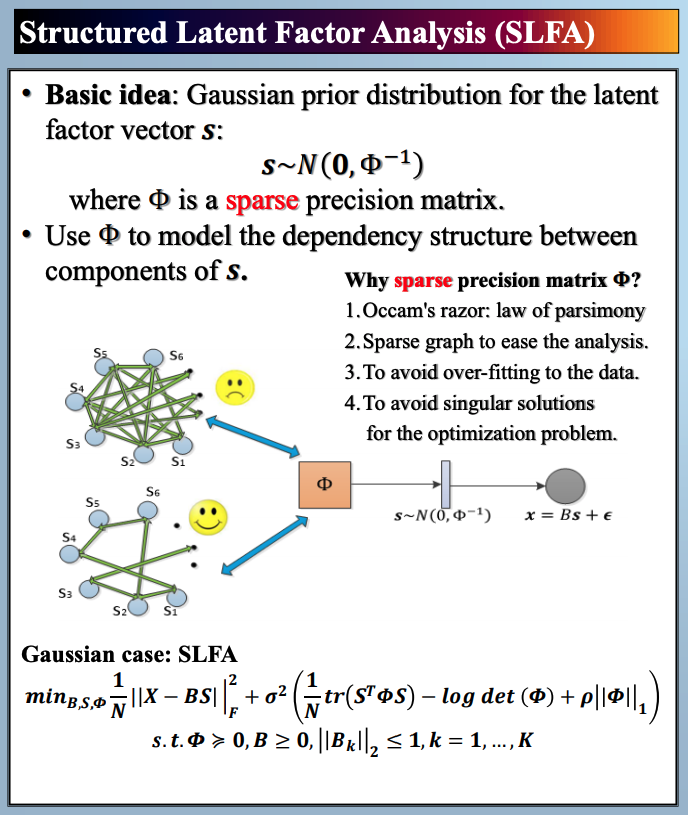
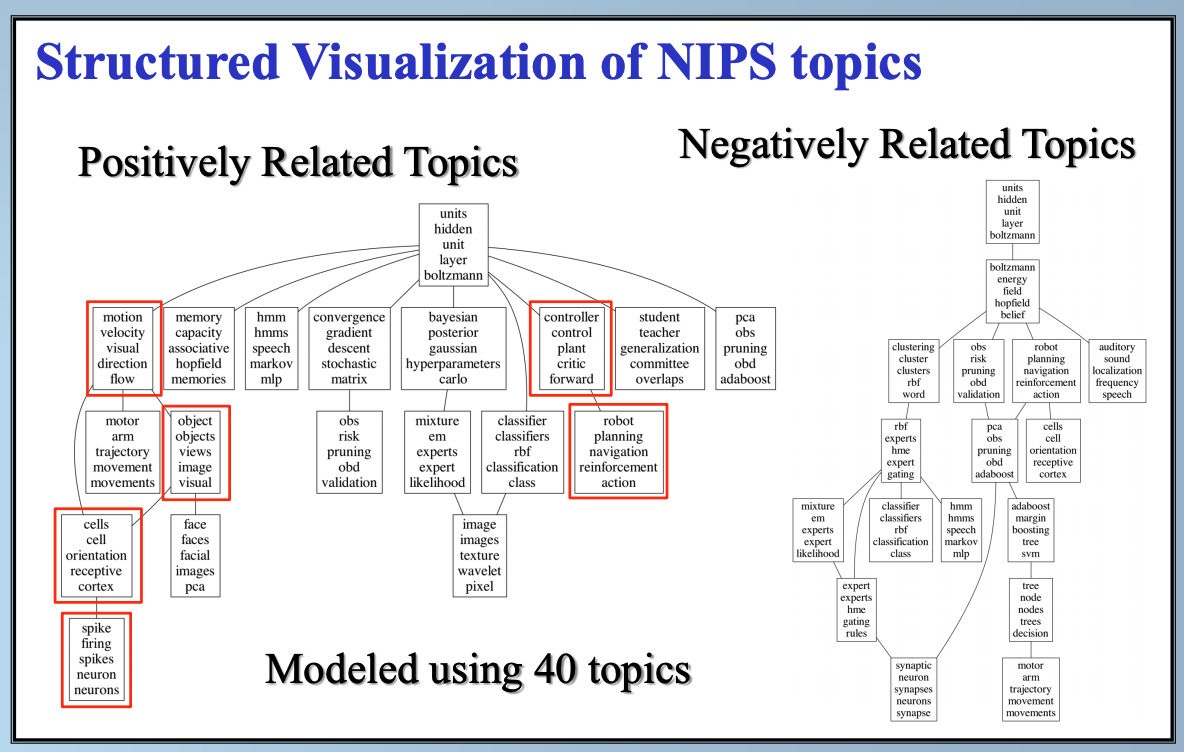

Citations
@inproceedings{he2012learning,
title={Learning the dependency structure of latent factors},
author={He, Yunlong and Qi, Yanjun and Kavukcuoglu, Koray and Park, Haesun},
booktitle={Advances in neural information processing systems},
pages={2366--2374},
year={2012}
}
Having trouble with our tools? Please contact Yanjun Qi and we’ll help you sort it out.
01 Feb 2009
Title: Semi-supervised multi-task learning for predicting interactions between HIV-1 and human proteins
- authors: Yanjun Qi, Oznur Tastan, Jaime G. Carbonell, Judith Klein-Seetharaman, Jason Weston
Abstract
-
Motivation: Protein–protein interactions (PPIs) are critical for virtually every biological function. Recently, researchers suggested to use supervised learning for the task of classifying pairs of proteins as interacting or not. However, its performance is largely restricted by the availability of truly interacting proteins (labeled). Meanwhile, there exists a considerable amount of protein pairs where an association appears between two partners, but not enough experimental evidence to support it as a direct interaction (partially labeled).
-
Results: We propose a semi-supervised multi-task framework for predicting PPIs from not only labeled, but also partially labeled reference sets. The basic idea is to perform multi-task learning on a supervised classification task and a semi-supervised auxiliary task. The supervised classifier trains a multi-layer perceptron network for PPI predictions from labeled examples. The semi-supervised auxiliary task shares network layers of the supervised classifier and trains with partially labeled examples. Semi-supervision could be utilized in multiple ways. We tried three approaches in this article, (i) classification (to distinguish partial positives with negatives); (ii) ranking (to rate partial positive more likely than negatives); (iii) embedding (to make data clusters get similar labels). We applied this framework to improve the identification of interacting pairs between HIV-1 and human proteins. Our method improved upon the state-of-the-art method for this task indicating the benefits of semi-supervised multi-task learning using auxiliary information.
Citations
@article{qi2010semi,
title={Semi-supervised multi-task learning for predicting interactions between HIV-1 and human proteins},
author={Qi, Yanjun and Tastan, Oznur and Carbonell, Jaime G and Klein-Seetharaman, Judith and Weston, Jason},
journal={Bioinformatics},
volume={26},
number={18},
pages={i645--i652},
year={2010},
publisher={Oxford University Press}
}


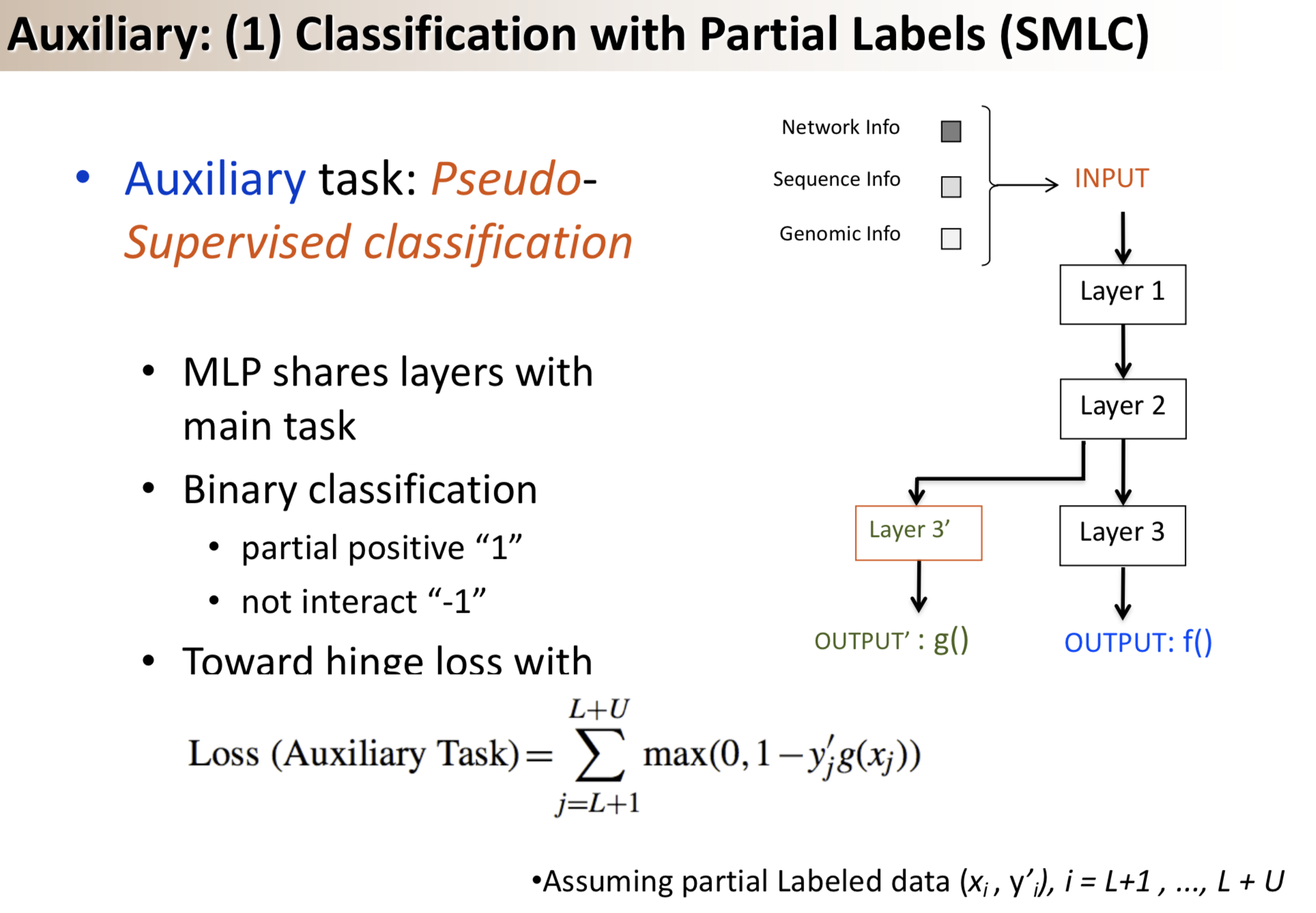
Having trouble with our tools? Please contact Yanjun Qi and we’ll help you sort it out.