EpiGenome (Index of Posts):
Our deep tools aim to extract knowledge from epigenomic signals, trying to figure out how they influence gene regulation..
This includes:
01 May 2020
Title: Graph Convolutional Networks for Epigenetic State Prediction Using Both Sequence and 3D Genome Data
Abstract
Motivation
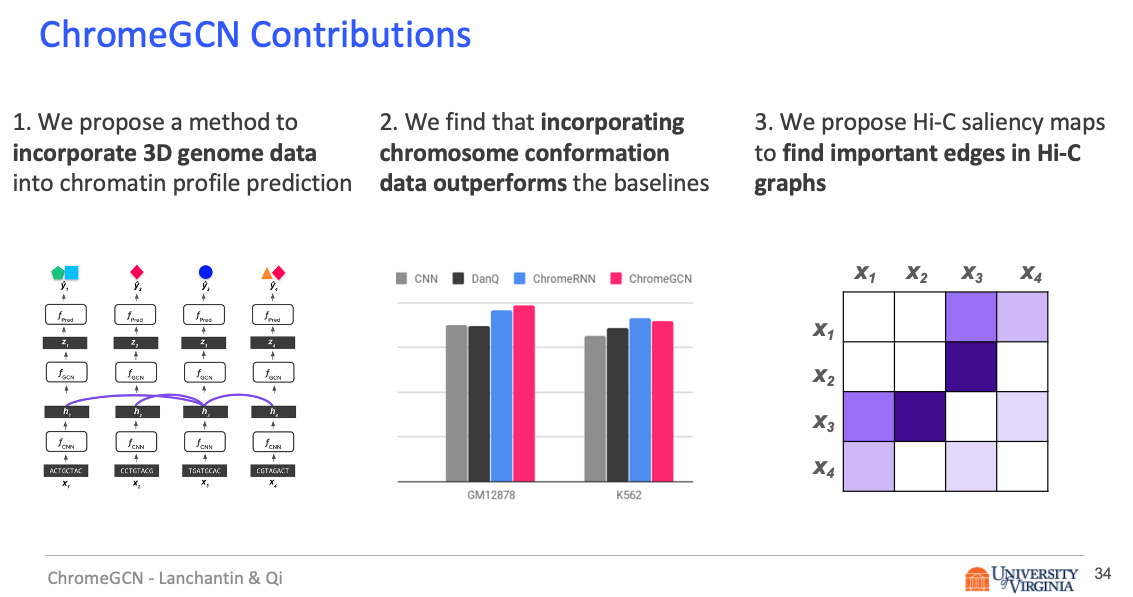
Predictive models of DNA chromatin profile (i.e. epigenetic state), such as transcription factor binding, are essential for understanding regulatory processes and developing gene therapies. It is known that the 3D genome, or spatial structure of DNA, is highly influential in the chromatin profile. Deep neural networks have achieved state of the art performance on chromatin profile prediction by using short windows of DNA sequences independently. These methods, however, ignore the long-range dependencies when predicting the chromatin profiles because modeling the 3D genome is challenging.
Results
In this work, we introduce ChromeGCN, a graph convolutional network for chromatin profile prediction by fusing both local sequence and long-range 3D genome information. By incorporating the 3D genome, we relax the independent and identically distributed assumption of local windows for a better representation of DNA. ChromeGCN explicitly incorporates known long-range interactions into the modeling, allowing us to identify and interpret those important long-range dependencies in influencing chromatin profiles. We show experimentally that by fusing sequential and 3D genome data using ChromeGCN, we get a significant improvement over the state-of-the-art deep learning methods as indicated by three metrics. Importantly, we show that ChromeGCN is particularly useful for identifying epigenetic effects in those DNA windows that have a high degree of interactions with other DNA windows.
Citations
@article{10.1093/bioinformatics/btaa793,
author = {Lanchantin, Jack and Qi, Yanjun},
title = "{Graph convolutional networks for epigenetic state prediction using both sequence and 3D genome data}",
journal = {Bioinformatics},
volume = {36},
number = {Supplement_2},
pages = {i659-i667},
year = {2020},
month = {12},
issn = {1367-4803},
doi = {10.1093/bioinformatics/btaa793},
url = {https://doi.org/10.1093/bioinformatics/btaa793},
eprint = {https://academic.oup.com/bioinformatics/article-pdf/36/Supplement\_2/i659/35336695/btaa793.pdf},
}
Having trouble with our tools? Please contact Jack and we’ll help you sort it out.
07 Sep 2018
Paper:
Abstract:
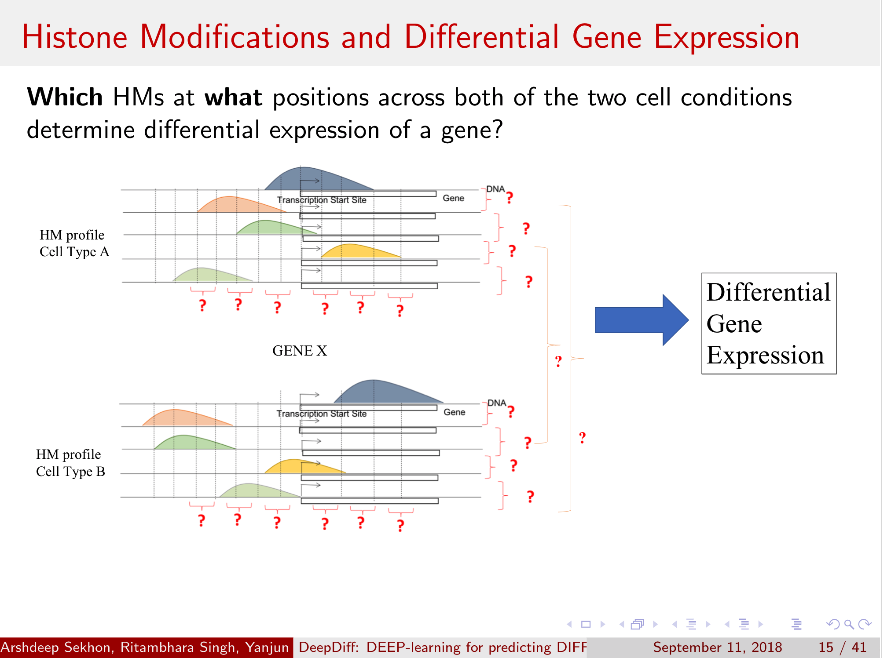
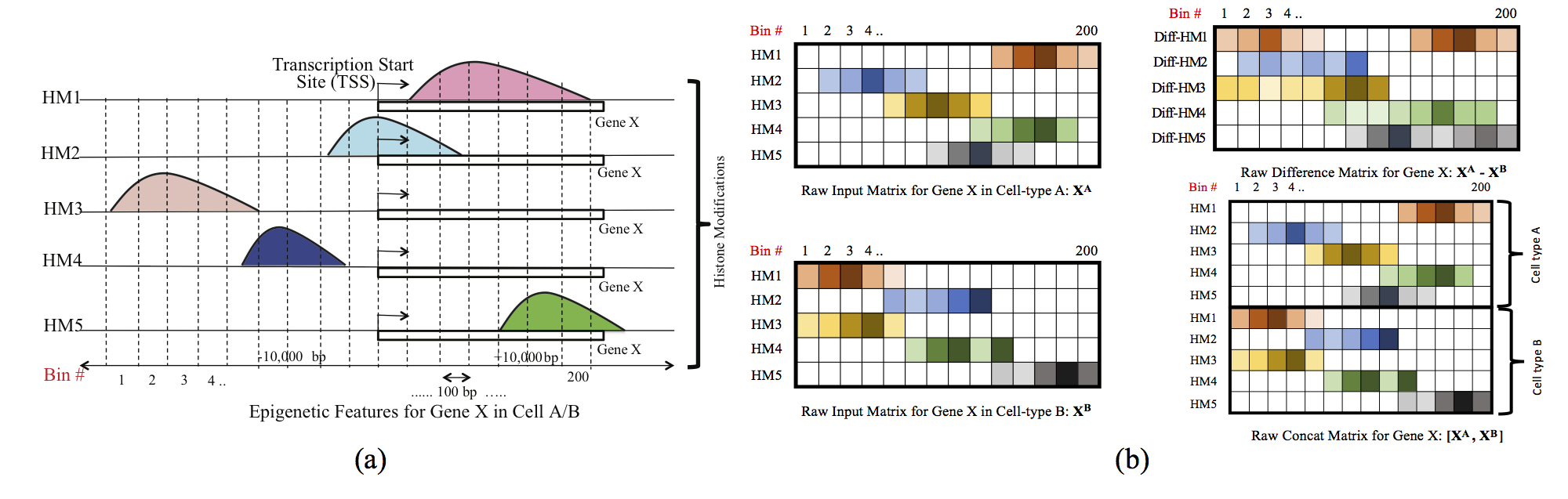
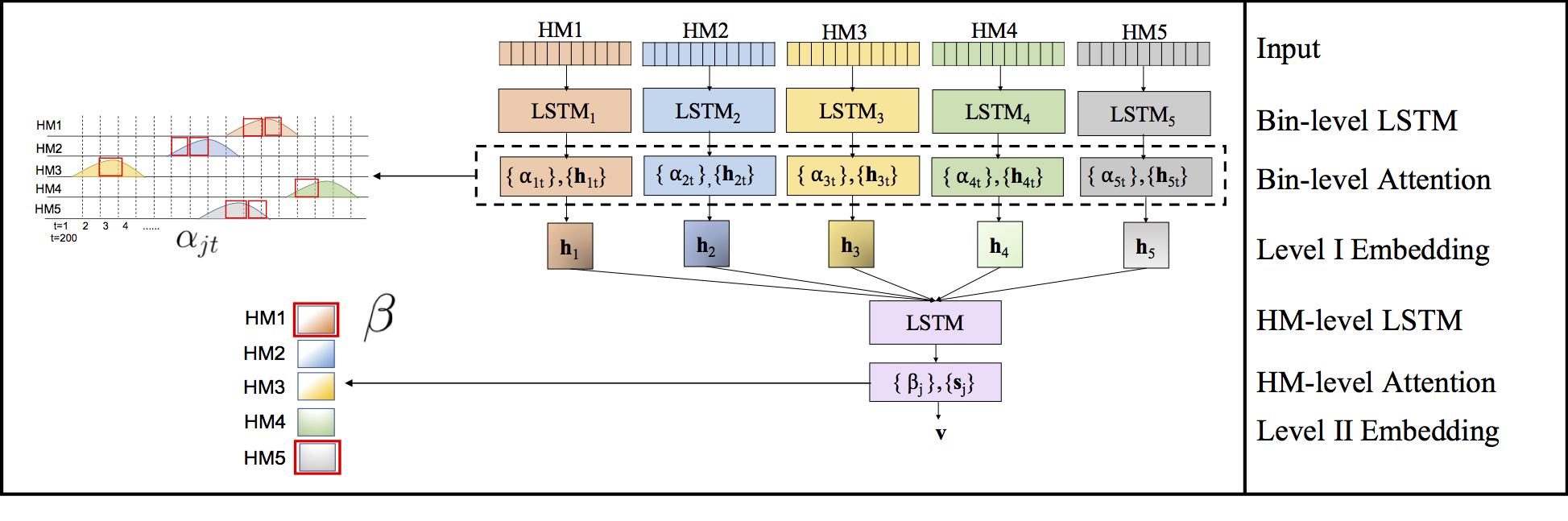
Computational methods that predict differential gene expression from histone modification signals are highly desirable for understanding how histone modifications control the functional heterogeneity of cells through influencing differential gene regulation. Recent studies either failed to capture combinatorial effects on differential prediction or primarily only focused on cell type-specific analysis. In this paper, we develop a novel attention-based deep learning architecture, DeepDiff, that provides a unified and end-to-end solution to model and to interpret how dependencies among histone modifications control the differential patterns of gene regulation. DeepDiff uses a hierarchy of multiple Long short-term memory (LSTM) modules to encode the spatial structure of input signals and to model how various histone modifications cooperate automatically. We introduce and train two levels of attention jointly with the target prediction, enabling DeepDiff to attend differentially to relevant modifications and to locate important genome positions for each modification. Additionally, DeepDiff introduces a novel deep-learning based multi-task formulation to use the cell-type-specific gene expression predictions as auxiliary tasks, encouraging richer feature embeddings in our primary task of differential expression prediction. Using data from Roadmap Epigenomics Project (REMC) for ten different pairs of cell types, we show that DeepDiff significantly outperforms the state-of-the-art baselines for differential gene expression prediction. The learned attention weights are validated by observations from previous studies about how epigenetic mechanisms connect to differential gene expression. Codes and results are available at deepchrome.org
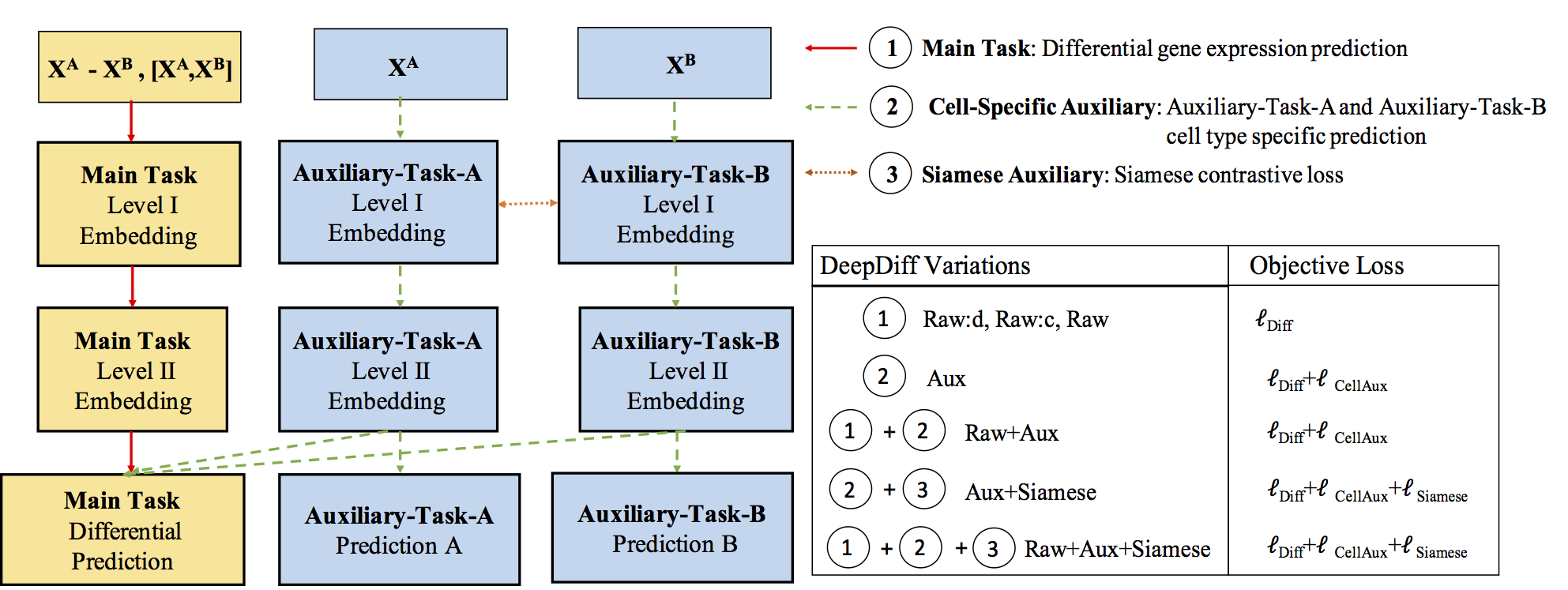
Citations
@article{ArDeepDiff18,
author = {Sekhon, Arshdeep and Singh, Ritambhara and Qi, Yanjun},
title = {DeepDiff: DEEP-learning for predicting DIFFerential gene expression from histone modifications},
journal = {Bioinformatics},
volume = {34},
number = {17},
pages = {i891-i900},
year = {2018},
doi = {10.1093/bioinformatics/bty612},
URL = {http://dx.doi.org/10.1093/bioinformatics/bty612},
eprint = {/oup/backfile/content_public/journal/bioinformatics/34/17/10.1093_bioinformatics_bty612/2/bty612.pdf}
}
Having trouble with our tools? Please contact Arsh and we’ll help you sort it out.
30 Jul 2017
Paper: @Arxiv + Published at [NIPS2017]
(https://papers.nips.cc/paper/7255-attend-and-predict-understanding-gene-regulation-by-selective-attention-on-chromatin.pdf)
Abstract:
The past decade has seen a revolution in genomic technologies that enable a flood of genome-wide profiling of chromatin marks. Recent literature tried to understand gene regulation by predicting gene expression from large-scale chromatin measurements. Two fundamental challenges exist for such learning tasks: (1) genome-wide chromatin signals are spatially structured, high-dimensional and highly modular; and (2) the core aim is to understand what are the relevant factors and how they work together? Previous studies either failed to model complex dependencies among input signals or relied on separate feature analysis to explain the decisions. This paper presents an attention-based deep learning approach; we call AttentiveChrome, that uses a unified architecture to model and to interpret dependencies among chromatin factors for controlling gene regulation. AttentiveChrome uses a hierarchy of multiple Long short-term memory (LSTM) modules to encode the input signals and to model how various chromatin marks cooperate automatically. AttentiveChrome trains two levels of attention jointly with the target prediction, enabling it to attend differentially to relevant marks and to locate important positions per mark. We evaluate the model across 56 different cell types (tasks) in human. Not only is the proposed architecture more accurate, but its attention scores also provide a better interpretation than state-of-the-art feature visualization methods such as saliency map.
Code and data are shared at www.deepchrome.net
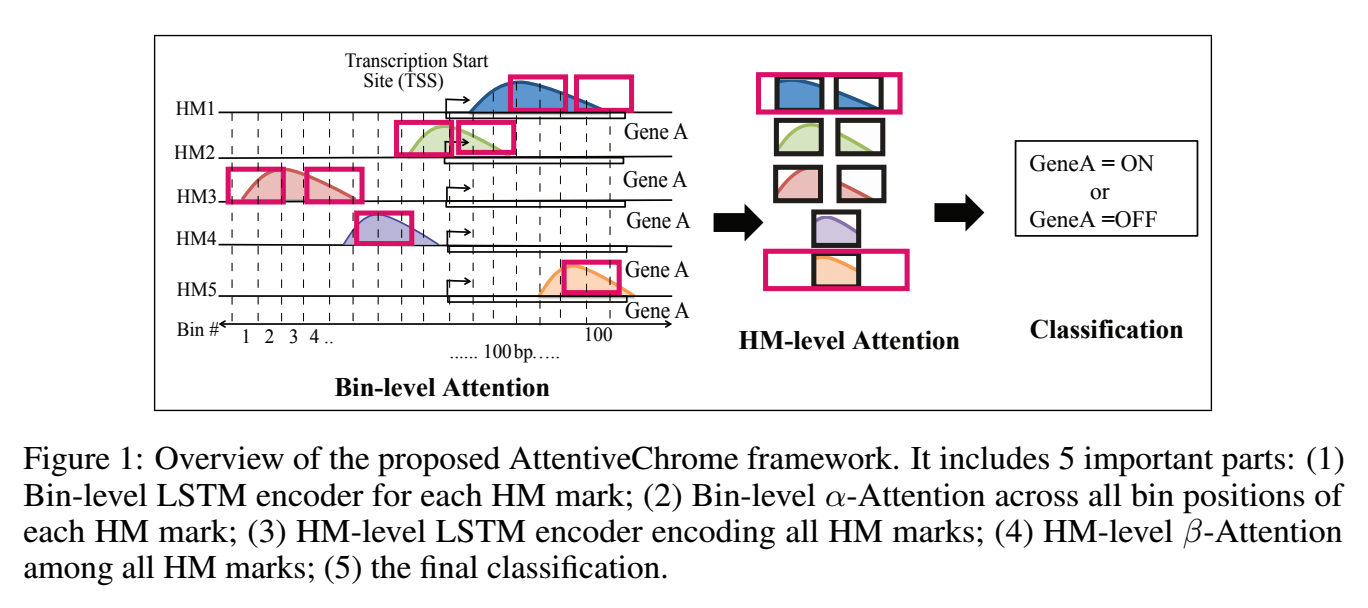
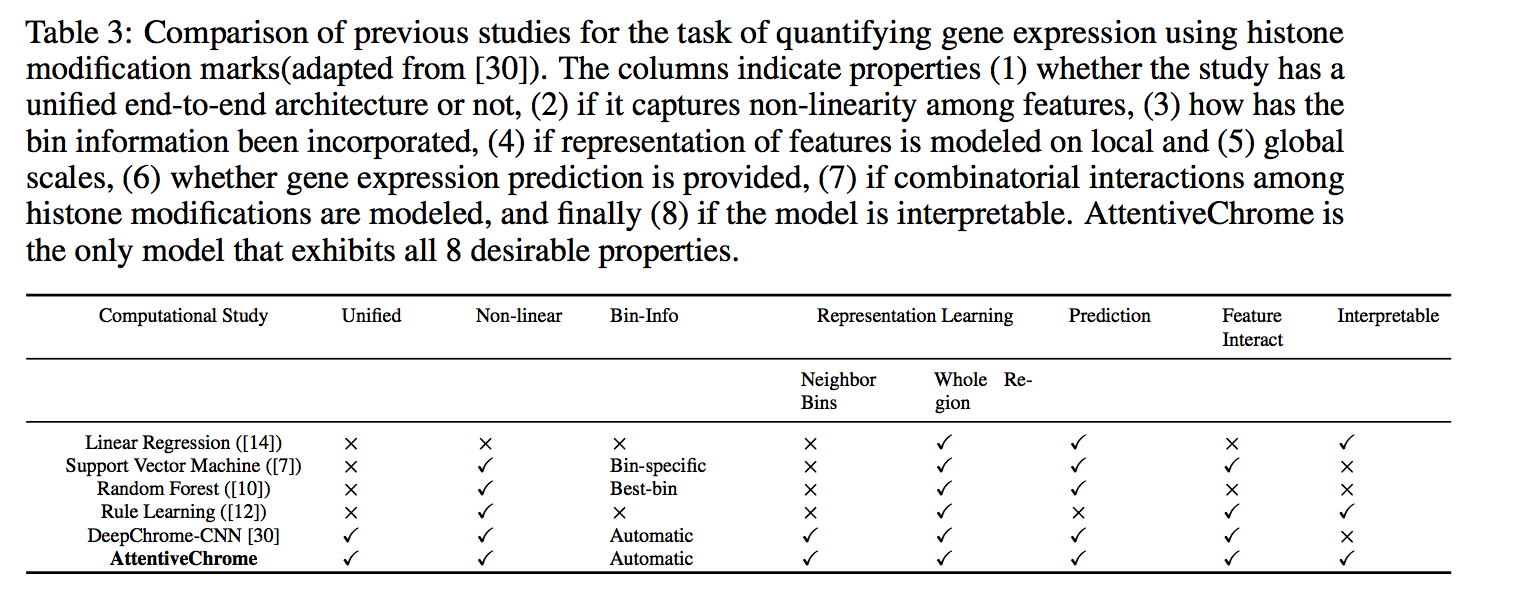
Citations
@inproceedings{singh2017attend,
title={Attend and Predict: Understanding Gene Regulation by Selective Attention on Chromatin},
author={Singh, Ritambhara and Lanchantin, Jack and Sekhon, Arshdeep and Qi, Yanjun},
booktitle={Advances in Neural Information Processing Systems},
pages={6769--6779},
year={2017}
}
Having trouble with our tools? Please contact Rita and we’ll help you sort it out.
10 Jun 2017
Abstract:
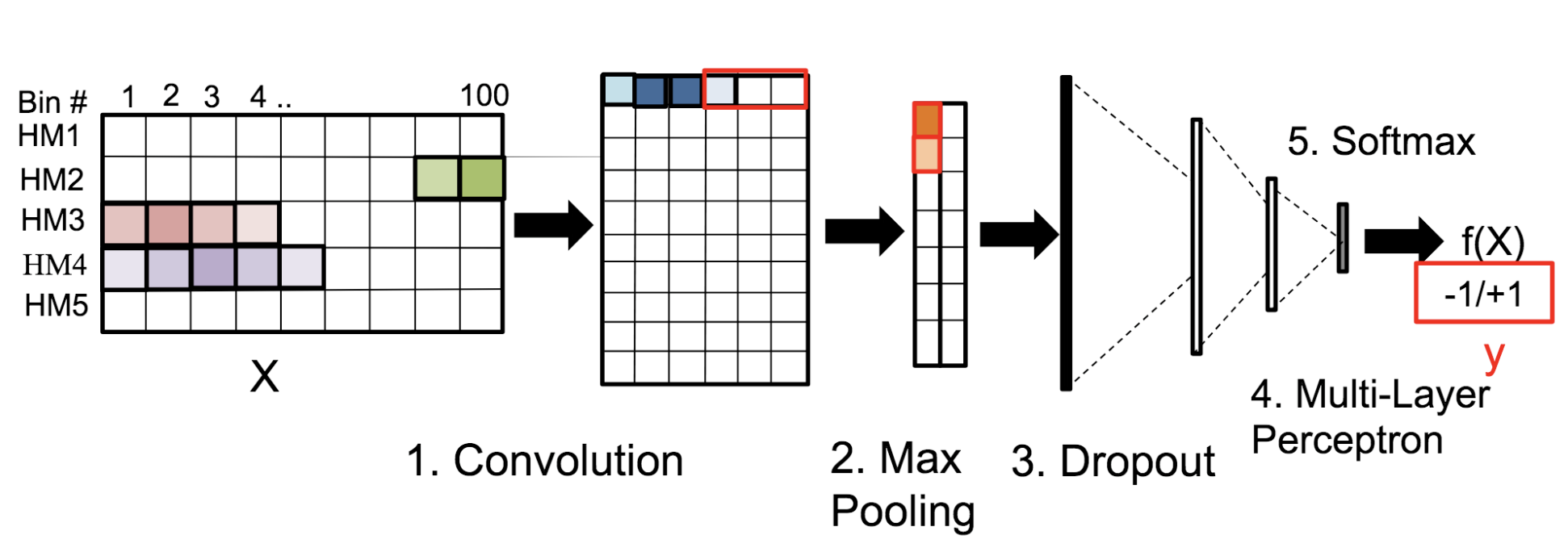
Motivation: Histone modifications are among the most important factors that control gene regulation. Computational methods that predict gene expression from histone modification signals are highly desirable for understanding their combinatorial effects in gene regulation. This knowledge can help in developing ‘epigenetic drugs’ for diseases like cancer. Previous studies for quantifying the relationship between histone modifications and gene expression levels either failed to capture combinatorial effects or relied on multiple methods that separate predictions and combinatorial analysis. This paper develops a unified discriminative framework using a deep convolutional neural network to classify gene expression using histone modification data as input. Our system, called DeepChrome, allows automatic extraction of complex interactions among important features. To simultaneously visualize the combinatorial interactions among histone modifications, we propose a novel optimization-based technique that generates feature pattern maps from the learnt deep model. This provides an intuitive description of underlying epigenetic mechanisms that regulate genes. Results: We show that DeepChrome outperforms state-of-the-art models like Support Vector Machines and Random Forests for gene expression classification task on 56 different cell-types from REMC database. The output of our visualization technique not only validates the previous observations but also allows novel insights about combinatorial interactions among histone modification marks, some of which have recently been observed by experimental studies.
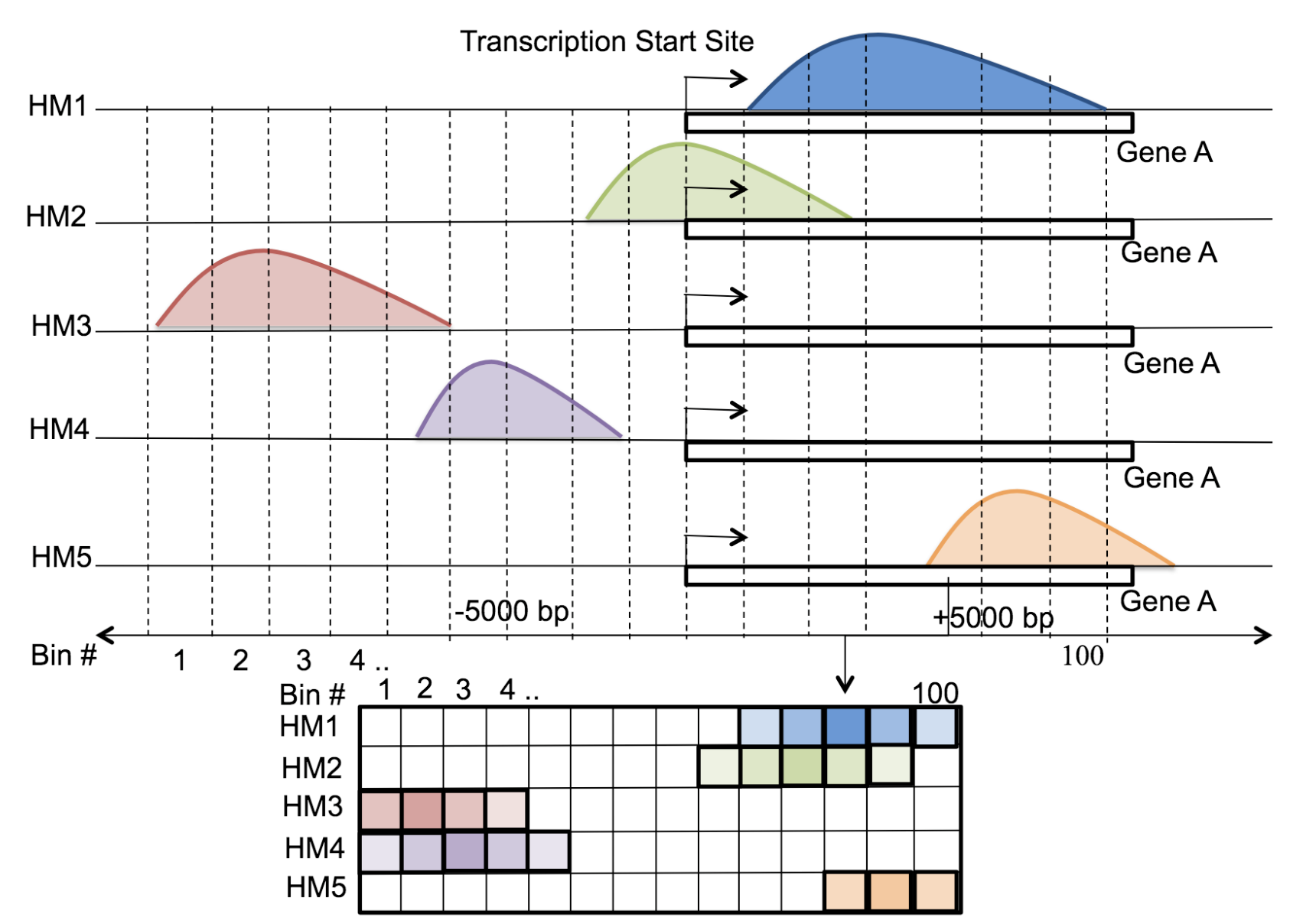
Citations
@article{singh2016deepchrome,
title={DeepChrome: deep-learning for predicting gene expression from histone modifications},
author={Singh, Ritambhara and Lanchantin, Jack and Robins, Gabriel and Qi, Yanjun},
journal={Bioinformatics},
volume={32},
number={17},
pages={i639--i648},
year={2016},
publisher={Oxford University Press}
}
Having trouble with our tools? Please contact Rita and we’ll help you sort it out.